Настройки выявления вариантов
Фильтрация ПЦР дубликатов#
После стадии выравнивания результаты подвергаются нескольким этапам дополнительной обработки. Это повышает чувствительность к низкочастотным вариантам, способствует более точному подсчету количества копий, который зависит от количества прочтений, а также ускоряет обработку образца. Если вы хотите сохранить все прочтения для дальнейшего анализа (например, если вы работаете с ампликонами), то вы можете отключить фильтрацию.
Пометка дубликатов и её отключение#
В процессе секвенирования количество оригинальных последовательностей фрагментированной ДНК увеличивается в полимеразной цепной реакции (ПЦР). Для дальнейшего анализа очень важно учесть дублирующиеся прочтения одного и того же фрагмента. Пометка дубликатов выполняется с помощью инструмента GATK MarkDuplicates. Этот инструмент находит в файле выравнивания повторяющиеся прочтения, определяет, какие из прочтений происходят от оригинальных последовательностей, а какие являются копиями (дубликатами), и помечает дубликаты. Дальнейший анализ ведется только по оригинальным прочтениям. Для отключения пометки дубликатов выключите соответствующий тумблер:

Ограничение покрытия сайта для поиска вариантов и его отключение#
Увеличение количества прочтений, приходящихся на сайт для поиска герминальных вариантов, после определенного порога
(50 прочтений на сайт у GATK) приводит к необоснованным вычислительным тратам и может снижать чувствительность
к некоторым типам вариантов. Ограничение покрытия сайта прочтения выполняется опцией
--max-reads-per-alignment-start инструмента для поиска герминальных вариантов
GATK HaplotypeCaller,
благодаря которой "лишние" прочтения случайным образом выбрасываются из анализа при превышении порога
на количество прочтений (50 прочтений). Для отключения ограничения покрытия сайта выключите соответствующий тумблер:

На заметку
Данная опция влияет только на поиск герминальных вариантов.
Фильтрация SNVs/Indels#
Фильтрация по capture kit и её отключение#
Как правило, для образцов, полученных в результате таргетного секвенирования, поиск вариантов проводится в тех же
геномных интервалах, в которых проводилось секвенирование. Таким образом, если для образца был определен
capture kit, то сначала с помощью
инструмента bedtools intersect
мы получаем интервалы пересечения этого capture kit с референсным геномом, а затем в этих интервалах осуществляется
поиск герминальных
(с помощью GATK HaplotypeCaller)
или соматических (с помощью
GATK Mutect2) вариантов.
Файл с интервалами указывается параметром -L (--intervals).
Для отключения фильтрации по capture kit выключите соответствующий тумблер:

Отбрасывание вариантов, не прошедших фильтрацию, и его отключение#
Выявленные коллером варианты обязательно проходят через стадию фильтрации, во время которой проверяется, удовлетворяет ли вариант порогам параметров фильтрации. В результате в поле FILTER аннотации проставляется значение "PASS", если вариант удовлетворяет всем параметрам фильтрации, или указывается тот параметр, порогу которого вариант не удовлетворяет. По умолчанию только варианты, прошедшие все фильтры, участвуют в дальнейшем анализе и отображаются в SNV Viewer. Вы можете отключить эту опцию - тогда в SNV Viewer и статистике Биоинформатического отчёта будут отображаться все варианты. Для этого выключите соответствующий тумблер:

На заметку
Данная опция влияет только на отображение не прошедших фильтрацию вариантов в SNV Viewer и Биоинформатическом отчёте. В дальнейшем анализе (например, в групповом) всё равно будут участвовать только фильтрованные варианты.
Выявление соматических SNVs/Indels#
Соматические мутации являются приобретенными и характерны для опухолевого процесса. Соответственно, выявление таких мутаций проводится только в образцах опухолевой ткани с использованием контроля (соответствующей неопухолевой ткани того же человека) или без контроля. Выявление соматических однонуклеотидных вариантов (single-nucleotide variants; SNVs) и коротких инсерций/делеций (indels) происходит с помощью инструмента GATK Mutect2, который работает в первую очередь путем сопоставления вариантов в опухоли и контроле (режим tumor-normal). Он также работает и для поиска вариантов в опухоли без контроля (режим tumor-only), но такой поиск отличается высоким уровнем ложных срабатываний (false positives).
Отключение поиска соматических SNVs/Indels#
Для отключения поиска соматических SNVs/Indels выключите соответствующий тумблер:

Выбор нормальной панели#
Нормальная панель (Panel of Normal; PON) - панель, полученная из образцов здоровой ткани (то есть ткани, не имеющей
соматических мутаций) для захвата повторяющихся технических артефактов с целью улучшения результатов
выявления соматических вариантов. При этом образцы здоровой ткани, на которых строится
панель, должны быть получены с помощью секвенирования со схожими техническими характеристиками, такими как тип
секвенирования, секвенатор, реагент Capture Kit и т.д. Таким образом, при выборе PON, подходящей для ваших данных,
необходимо руководствоваться техническими характеристиками секвенирования образца опухолевой ткани. Чем
точнее подобрана панель, тем более специфичным становится анализ и может быть выявлено больше вариантов.
Панель предоставляется инструменту
GATK Mutect2
с помощью параметра --panel-of-normals (-pon).
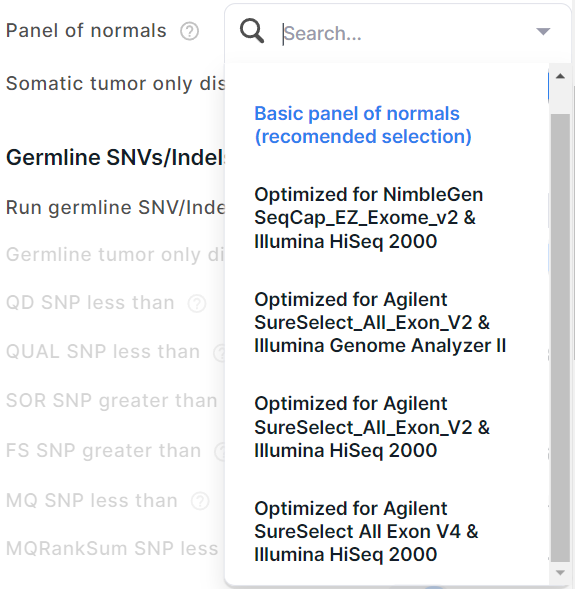
Для выбора доступны следующие нормальные панели:
- Basic panel of normals - панель, полученная из 110 образцов, случайно выбранных из 181 образца здоровой ткани человека. Панель показала самые высокие значения чувствительности и специфичности по сравнению с другими панелями при выявлении соматических SNVs/Indels в образце из золотого стандарта (образец секвенирования рака молочной железы SRR7890918 и образец крови SRR7890919). Выбрана по умолчанию.
- Optimized for NimbleGen SeqCap_EZ_Exome_v2 & Illumina HiSeq 2000 - панель, полученная для 30 образцов секвенирования на платформе Illumina HiSeq 2000 с реагентом Capture Kit NimbleGen SeqCap_EZ_Exome_v2.
- Optimized for Agilent SureSelect_All_Exon_V2 & Illumina Genome Analyzer II - панель, полученная для 30 образцов секвенирования на платформе Illumina Genome Analyzer II с реагентом Capture Kit Agilent SureSelect_All_Exon_V2.
- Optimized for Agilent SureSelect_All_Exon_V2 & Illumina HiSeq 2000 - панель, полученная для 30 образцов секвенирования на платформе Illumina HiSeq 2000 с реагентом Capture Kit Agilent SureSelect_All_Exon_V2.
- Optimized for Agilent SureSelect All Exon V4 & Illumina HiSeq 2000 - панель, полученная для 30 образцов секвенирования на платформе Illumina HiSeq 2000 с реагентом Capture Kit Agilent SureSelect All Exon V4.
- None - выберите эту опцию, если хотите провести выявление соматических SNVs/Indels без нормальной панели.
Для смены нормальной панели для выявления соматических SNVs/Indels нажмите на значение поля "Panel of normals" и выберите нужную панель:
Отключение поиска соматических SNVs/Indels у одиночного образца опухолевой ткани#
Для отключения поиска соматических SNVs/Indels у одиночного образца опухолевой ткани (режим tumor-only) выключите соответствующий тумблер:

На заметку
Отключение не затрагивает пайплайн анализа наборов образцов, включающих образец неопухолевой ткани (normal).
Выявление герминальных SNVs/Indels#
Герминальные мутации являются наследуемыми и могут быть обнаружены в любых клетках организма. Выявляние таких мутаций проводится для одиночных образцов опухолевой ткани без контроля и для образцов неопухолевой ткани. Выявление герминальных однонуклеотидных вариантов (single-nucleotide variants; SNVs) и коротких инсерций/делеций (indels) происходит с помощью инструмента GATK HaplotypeCaller.
Отключение поиска герминальных SNVs/Indels#
Для отключения поиска герминальных SNVs/Indels выключите соответствующий тумблер:

Отключение поиска герминальных SNVs/Indels у одиночного образца опухолевой ткани#
Для отключения поиска герминальных SNVs/Indels у одиночного образца опухолевой ткани выключите соответствующий тумблер:

На заметку
Отключение не затрагивает пайплайн анализа наборов образцов, включающих образец неопухолевой ткани (normal).
Настройка объединения фазовых групп#
Фазовая группа - это объединение гомозиготных вариантов, расположенных в пределах определенного геномного интервала (окна размера N).
Для отключения объединения вариантов в фазовые группы выключите соответствующий тумблер:

Чтобы настроить размеры геномного интервала, в пределах которого гомозиготы будут объединены в фазовую группу, нажмите на текущее значение (размер окна N = 3 п.н. по умолчанию) и отредактируйте (минимальное значение = 1, максимальное значение = 10). Для сохранения изменений нажмите клавишу "Enter" или кликните левой кнопкой мыши в свободное место на странице.
Параметры хардфильтрации (hard filtering) герминальных SNVs/Indels#
Нажмите, чтобы прочитать о параметрах хардфильтрации и их порогах
| Параметр фильтрации | Описание параметра | Значение порога параметра по умолчанию |
| QD (QualByDepth) | Достоверность варианта (значение из поля QUAL), нормализованная по нефильтрованнной глубине аллеля (AD) варианта в выборке вариантов с генотипом, отличным от hom-ref (homozygous/reference; генотип, при котором оба аллеля являются референсными). Для фильтрации лучше использовать QD, а не QUAL или фильтрованную глубину (DP) напрямую. Поскольку каждое прочтение вносит небольшой вклад в оценку QUAL, варианты в областях с глубоким покрытием могут иметь искусственно завышенные оценки QUAL, создавая впечатление, что вызов (call) варианта подтверждается большим количеством доказательств, чем это есть на самом деле. Чтобы этого избежать, необходимо нормализовать достоверность вариантов по глубине, что дает более объективную картину того, насколько хорошо поддерживается вызов (call). | QD < 2 |
| QUAL (качество варианта) | Вероятность существования (по шкале Phred) варианта в этом сайте с учетом данных секвенирования. | QUAL < 30 |
| SOR (StrandOddsRatio) | Смещение цепей, оцененное с помощью симметричного теста отношения шансов. Смещение цепей - это тип смещения последовательности, при котором предпочтение отдается одной цепи ДНК перед другой, что может привести к неправильной оценке количества доказательств, наблюдаемых для одного аллеля по сравнению с другим. StrandOddsRatio оценивает наличие смещения между прямыми и обратными цепями для референсного или альтернативного аллеля (аллелей). | SOR > 3 для SNVs (выключен для indels) |
| FS (FisherStrand) | Вероятность смещения цепей на сайте, оцененная с помощью точного критерия Фишера. Смещение цепей - это тип смещения последовательности, при котором предпочтение отдается одной цепи ДНК перед другой, что может привести к неправильной оценке количества доказательств, наблюдаемых для одного аллеля по сравнению с другим. FisherStrand определяет, существует ли смещение цепи между прямой и обратной цепями для референсного или альтернативного аллеля (то есть встречается ли альтернативный аллель чаще или реже на прямой или обратной цепи, чем референсный аллель). Результат FisherStrand представляет собой p-значение по шкале Phred. Чем выше это значение, тем больше вероятность смещения. Большее смещение свидетельствует о ложноположительных вызовах (false positive calls) вариантов. FisherStrand хуже учитывает большие объемы данных в ситуациях с высоким покрытием по сравнению с StrandOddsRatio. Кроме того, FisherStrand имеет тенденцию "штрафовать" варианты, которые встречаются на концах экзонов. Варианты на концах экзонов, как правило, покрываются прочтениями только в одном направлении цепи, и FisherStrand дает таким вариантам плохую оценку, тогда как StrandOddsRatio учитывает соотношение прочтений, охватывающих оба аллеля. | FS > 60 для SNVs, FS > 200 для indels |
| MQ (RMMSMappingQuality) | Среднеквадратичное значение качества сопоставления прочтений по всем образцам. Представляет собой оценку общего качества картирования прочтений, покрывающих сайт варианта, усредненную по всем образцам в когорте, т.е. квадратный корень среднего квадрата качества картирования сайта варианта. Таким образом, оценка учитывает стандартное отклонение качества картирования. Низкое стандратное отклонение означает, что все значения близки к среднему, тогда как высокое стандартное отклонение означает, что все значения далеки от среднего. Когда качество картирования на сайте хорошее, MQ будет около 60. | MQ < 40 для SNVs (выключен для indels) |
| MQRankSum (MappingQualityRankSumTest) | Тест суммы рангов для сопоставления качества картирования прочтения REF и качества картирования прочтения ALT. MQRankSum на уровне варианта сравнивает качество картирования прочтений, поддерживающих референсный аллель, с прочтениями, поддерживающими альтернативный аллель. Наилучшим результатом является значение, близкое к нулю, что указывает на то, что разница практически отсутствует. Отрицательное значение указывает на то, что прочтения, поддерживающие альтернативный аллель, имеют более низкие показатели качества картирования, чем прочтения, поддерживающие референсный аллель, и наоборот: положительное значение указывает на то, что прочтения, поддерживающие альтернативный аллель, имеют более высокие показатели качества картирования, чем прочтения, поддерживающие референсный аллель. Обнаружение любой статистически значимой разницы в качестве предполагает, что процесс секвенирования и/или картирования мог быть искажен артефактом. На практике при оценке качества варианта отфильтровываются только низкие отрицательные значения, поскольку идея состоит в том, чтобы отфильтровать варианты, для которых качество данных, подтверждающих альтернативный аллель, сравнительно низкое. Обратный случай, когда качество данных, поддерживающих референсный аллель, ниже, не очень информативен для фильтрации вариантов. | MQRankSum < -12.5 для SNVs (выключен для indels) |
| ReadPosRankSum (ReadPosRankSumTest) | Тест суммы рангов для относительных позиций аллелей REF и ALT в прочтениях. ReadPosRankSum на уровне варианта проверяет, различаются ли положения референсного и альтернативного аллеля в прочтениях, которые поддерживают вариант. Нахождение аллеля только вблизи концов прочтений указывает на ошибку, потому что именно там секвенаторы склонны делать больше всего ошибок. Однако некоторые варианты, расположенные вблизи краев областей секвенирования, обязательно будут перекрыты концами прочтений, поэтому нельзя просто установить абсолютный порог «минимального расстояния от конца считывания». Вот почему полезен тест суммы рангов, чтобы оценить, есть ли разница в том, насколько хорошо поддерживаются референсный и альтернативный аллели. Наилучшим результатом является значение, близкое к нулю, поскольку оно указывает на небольшую разницу между позициями референсного и альтернативного аллелей в прочтениях. Отрицательное значение говорит о том, что альтернативный аллель встречается на концах прочтений чаще, чем референсный аллель, и наоборот: положительное значение указывает на то, что референсный аллель встречается на концах прочтений чаще, чем альтернативный. Обнаружение статистически значимой разницы в относительном положении в любом случае указывает на то, что процесс секвенирования мог быть искажен артефактом. На практике при оценке качества варианта отфильтровываются только низкие отрицательные значения, поскольку идея состоит в том, чтобы отфильтровать варианты, для которых качество данных, подтверждающих альтернативный аллель, сравнительно низкое. Обратный случай, когда качество данных, поддерживающих эталонный аллель, ниже, не очень информативен для фильтрации вариантов. | ReadPosRankSum < -8 для SNVs; ReadPosRankSum < -20 для indels |
Если хотя бы один из параметров хардфильтрации варианта удовлетворяет установленному порогу, то такой вариант не проходит фильтрацию.
Отключение или включение параметров хардфильтрации и изменение их порогов#
Чтобы изменить значение порога параметра хардфильтрации, нажмите на текущее значение и отредактируйте. Для сохранения изменений нажмите клавишу "Enter" или кликните левой кнопкой мыши в свободное место на странице.
Чтобы отключить или включить параметр хардфильтрации, выключите или включите соответствующий тумблер. Включенный
тумблер имеет синий цвет и смещён вправо: ![]() , а выключенный имеет серый цвет и смещён влево:
, а выключенный имеет серый цвет и смещён влево: ![]() .
.
Включение фильтрации герминальных SNVs/Indels с помощью CNN#
Помимо хардфильтрации для герминальных вариантов применяется фильтрация на основе оценок предварительно обученной сверточной нейронной сети (convolutional neural network; CNN) 2D, реализованной в GATK CNNScoreVariants. Фильтрация с помощью CNN занимает много времени и не вносит большой вклад в фильтрацию по сравнению с хардфильтрацией. Подробнее о вкладке фильтрации с помощью CNN можно узнать здесь.
Для включения фильтрации герминальных SNVs/Indels с помощью CNN модели включите соответствующий тумблер:

Выявление структурных вариаций#
Структурные вариации (structural variation; SV) - длинные делеции, тандемные дупликации, инверсии и транслокации. Выявляются с помощью инструмента Delly1 - интегрированного метода прогнозирования геномных перестроек по всему геному, который может выявлять, генотипировать и визуализировать структурные вариации с разрешением до одного нуклеотида в данных массивно-параллельного секвенирования с короткими прочтениями. Для пары образцов опухоль/контроль применяется пайплайн с нормальным образцом в качестве контроля. Для одиночного образца неопухолевой ткани или одиночного образца опухолевой ткани применяется пайплайн без контрольного образца. При этом результатом выявления структурных вариаций у одиночного образца опухолевой ткани является смесь соматических и герминальных SV из-за отсутствия соответствующего контрольного образца.
Отключение поиска структурных вариаций#
Для отключения поиска структурных вариаций у образца выключите соответствующий тумблер:

Выявление вариации числа копий#
Вариация числа копий (copy number variation; CNV) - это явление, при котором в геноме наблюдается вариабельное количество фрагментов ДНК. Длина таких фрагментов обычно колеблется от 1 тыс. до нескольких млн пар оснований. CNV возникают в результате делеций и дупликаций и делятся на две основные группы: уменьшение числа копий и увеличение числа копий в геноме. Такие удаленные или дублированные фрагменты ДНК могут понизить или повысить число копий определенного гена, что, в свою очередь, влияет на экспрессию продукта этого гена - белка или некодирующей РНК. Вариации числа копий имеют большое значение при синдромах и раке. Выводы о количестве копий делаются на основе различий в глубине секвенирования (количестве прочтений) данных секвенирования. Выявление вариации числа копий происходит с помощью инструмента CNVkit2, который умеет определять и визулизировать число копий как в данных таргетного секвенирования, так и в данных полноэкзомного секвенирования. Для пары образцов опухоль/контроль образец опухоли нормализуется по отношению к соответствующему контролю. Для одиночных образцов опухолевой или неопухолевой ткани нормализация на основе контроля не применяется. Также для одиночного образца опухолевой ткани вариации не разделяются на соматические и герминальные из-за отсутствия соответствующего контрольного образца.
Отключение поиска вариации числа копий#
Для отключения поиска вариации числа копий у образца выключите соответствующий тумблер:

Порог вызова CNV#
Пороговое значение log2FC, ниже которого вызывается одна аутосома вместо двух в норме. log2FC - это логарифмическое соотношение выявленного числа копий к нормальному числу копий (равно двум для аутосом и X-хромосомы в случае генотипа XX либо единице для половых хромосом в случае генотипа XY). На основе выбранного порогового значения рассчитываются пороги для вызова CNV. На примере значения порога по умолчанию (-0,7) разберём, какими будут пороги для вызова делеций и дупликаций на аутосомах и половых хромосомах:
- Вариации числа копий на аутосомах с Log2FC ≤ -0.7 принимаются за делеции (то есть ниже этого порога вызывается одна аутосома вместо двух в норме).
- Вариации числа копий на X- и Y-хромосомах с Log2FC ≤ -2.11 принимаются за делеции.
- Вариации числа копий на аутосомах с Log2FC ≥ 0.46 принимаются за дупликации. Порог для дупликации на аутосоме рассчитывается по следующей формуле: Log2FC = log2((2+D)/2), где D - "количество" копийности, которое "теряется" при Log2FC=-0.7, а 2 - количество сестринских хроматид в аутосоме.
- Вариации числа копий на X- и Y-хромосомах с Log2FC ≥ 0.82 принимаются за дупликации.
- Вариации, не удовлетворяющие порогам, т.е. вариации на аутосомах с -0.7 < Log2FC < 0.46 и вариации на половых хромосомах с -2.11 < Log2FC < 0.82, не проходят фильтрацию.
Нажмите, чтобы посмотреть, как производится расчёт порогов log2FC для делеций и дупликаций на аутосомах и половых хромосомах
Дельта числа копий: D = 2 – C = 2 – 2 × 2T21.
Тогда соответствующий порог вызова трёх копий вместо двух на аутосомах вычисляется так: T23 = log2((2+D)/2) = log2(2-2T21).
Порог для вызова делеций на аутосомах: lo = T21.
Порог для вызова дупликаций на аутосомах: hi = T23.
Для вычисления порога для вызова трёх копий вместо двух на Х-хромосоме нормализуем генотип XY, поделив его на число копий, равное 1: T23X= log2((2+D)/1) = log2(2+2-2×2T21) = 1 + log2(2-2T21) = 1 + T23.
Вычисление порога для вызова дупликаций на X-хромосоме:
- если нет Y-хромосомы: Xhi = 1 + T23;
- если есть Y-хромосома: Xhi = log2(3-2×2T21).
Порог для вызова одной копии вместо двух на Х-хромосоме: T21X = log2((2-D)/1) = log2(2-2+2×2T21) = 1 + T21.
Порог для вызова ни одной копии вместо одной на X-хромосоме: T10X = log2((1-D)/1) = log2(1-2+2×2T21) = log2(-1+2×2T21).
Вычисление порога для вызова делеций на X-хромосоме:
- если нет Y-хромосомы: Xlo = 1 + T21;
- если есть Y-хромосома: Xlo = log2(-1+2×2T21).
Порог для вызова одной копии вместо ни одной на Y-хромосоме в случае генотипа XX: T01 = log2((0+D)/1) = log2(2-2×2T21) = log2(2×(1-2T21)) = 1 + log2(1-2T21).
Вычисление порога для вызова дупликаций Yhi и делеций Ylo на Y-хромосоме:
- если нет Y-хромосомы: Yhi = 1 + log2(1-2T21)); Ylo = -∞;
- если есть Y-хромосома: Yhi = Xhi; Ylo = Xlo.
Поиск только герминальных анеуплоидий#
По умолчанию для образца неопухолевой ткани проводится поиск вариации числа копий как целых хромосом, так и их плечей (q и p). Если вы хотите выявить только герминальные анеуплоидии (вариации числа копий хромосом), включите соответствующий тумблер:

Выбор панели референсных образцов для WGS#
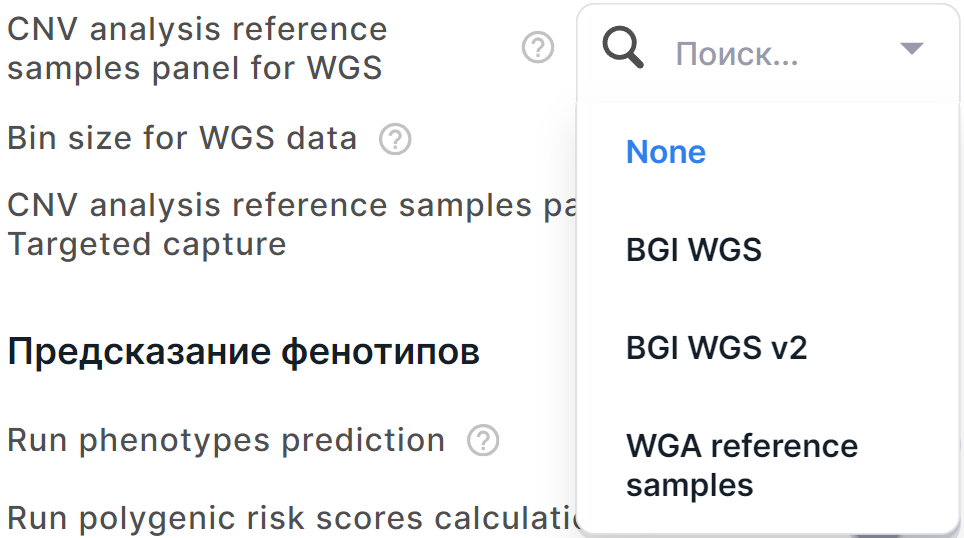
Анализ CNV более эффективен, когда для нормализации используется панель референсных образцов. По умолчанию панель не выбрана (None, т.е. нормализация отключена). Для выбора доступны следующие панели:
- BGI WGS - панель на основе WGS образцов, которые секвенировались на BGI. Панель рассчитана для бинов следующих размеров: 3000, 5000, 100000.
- BGI WGS v2 - панель на основе WGS образцов, которые секвенировались на BGI. Панель рассчитана для бина размером 3000.
- WGA reference samples - панель, которая позволяет проводить анализ CNV для результатов полногеномного секвенирования с низким покрытием (low-pass WGS). Панель рассчитана для бинов следующих размеров: 100000, 300000, 500000, 1000000, 2000000, 5000000.
Для выбора панели нажмите на значение поля "CNV analysis reference samples panel for WGS" и выберите нужную панель:
Если тип секвенирования образца автоматически определяется как направленный отбор, то выбранная референсная панель CNV игнорируется.
Размер бина для WGS#
Бин (bin) - это интервал, в котором проводится подсчет количества прочтений. Если у вас данные с низким покрытием, выбирайте высокие значения среднего размера бина (например, до 500,000 для low-pass WGS), а если данные с высоким покрытием, то выбирайте низкие значения (например, 1000 для данных с покрытием 30X). По умолчанию средний размер бина = 100,000. Чтобы его поменять, нажмите на текущее значение и отредактируйте. Минимальное допустимое значение бина = 1000, максимальное = 5,000,000. Для сохранения изменений нажмите клавишу "Enter" или кликните левой кнопкой мыши в свободное место на странице. Если вы выбрали панель референсных образцов для WGS, то выберите размер бина, для которого она была рассчитана, из списка.
Выбор панели референсных образцов для направленного отбора#
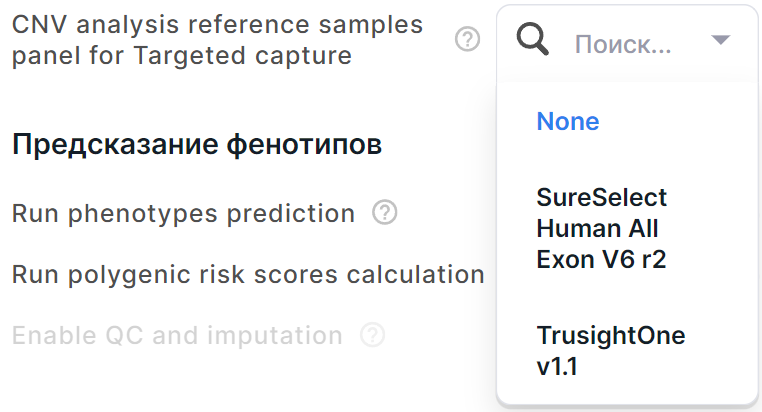
Анализ CNV более эффективен, когда для нормализации используется панель референсных образцов. Выбранная референсная панель CNV должна соответствовать реагенту capture kit, использованному для приготовления анализируемого образца. По умолчанию панель не выбрана (None, т.е. нормализация отключена). Для выбора доступны следующие панели:
- SureSelect Human All Exon V6 r2 - панель для выявления CNV в образцах, полученных в результате таргетного секвенирования с реагентом capture kit "SureSelect Human All Exon V6 r2".
- TrusightOne v1.1 - панель для выявления CNV в образцах, полученных в результате таргетного секвенирования с реагентом capture kit "TruSight One v1.1".
Все референсные панели CNV для направленного отбора рассчитаны только для одного разрешения, поэтому на анализ не будет влиять выбранный размер бина.
Для выбора панели нажмите на значение поля "CNV analysis reference samples panel for Targeted capture" и выберите нужную панель:
Если тип секвенирования образца автоматически определяется как WGS, то выбранная референсная панель CNV игнорируется.
Предсказание фенотипов#
Предсказание фенотипов происходит по однонуклеотидным полиморфизмам (single nucleotide polymorphism; SNP), ассоциированным с определенным проявлением признака согласно моделям, основанным на мультиномиальной логистической регрессии (multinomial logistic regression; MLR). Мы используем модели, позволяющие предсказать такие фенотипические признаки, как цвет волос, цвет глаз, цвет кожи, веснушчатость, непереносимость лактозы, группа крови, метаболизм алкоголя, запах тела (подмышечный осмидроз), тип ушной серы, чувствительность рецепторов к горькому вкусу, пол и фармакокинетика некоторых препаратов. Предсказание фенотипов можно провести для данных генотипирования в формате GT или аннотированных герминальных SNVs/Indels в формате VCF.
Включение предсказания фенотипов#
Для включения предсказания фенотипов у образца включите соответствующий тумблер:

Отключение контроля качества и импутации#
Пайплайн предсказания фенотипов включает три подготовительных шага перед самим предсказанием. Первый - фильтрация SNP по качеству - осуществляется с помощью инструмента PLINK. Затем происходит импутация (определение негенотипированных маркеров) c помощью bcftools и Beagle3. Импутация значительно увеличивает количество маркеров, на основе которых будет происходить предсказание фенотипов. И в конце снова повторяется фильтрация маркеров по качеству. Эти шаги важны для качества предсказания фенотипов, но не всегда завершаются успешно для аннотированных вариантов в формате VCF. Для отключения контроля качества и импутации выключите соответствующий тумблер:

- Rausch T., Zichner T., Schlattl A. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28(18), i333:i339 (2012)↩
- Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: Genome-wide copy number detection and visualization from targeted sequencing. PLOS Computational Biology 12(4), e1004873 (2014)↩
- Browning B.L., Zhou Y., Browning S.R. A one-penny imputed genome from next generation reference panels. Am J Hum Genet 103(3), 338-348 (2018)↩