Отчёт на основе фенотипа ClinVar
Отчёт включает состояние (фенотип ClinVar), риск развития которого у пациента высок в связи с обнаруженными в образце пациента патогенными или вероятно патогенными вариантами, ассоциированными с данным состоянием, и рекомендации, которые могут снизить риски возникновения этого состояния.
Чтобы получить такой отчёт для образца, создайте шаблон отчёта, включающий блок "SNVs/Indels по фенотипам ClinVar".
Отчёт разбит на три основных раздела: Результат отчёта и общая рекомендация, Подробный отчёт и Исключенные варианты.
1. Результат отчёта и общая рекомендация#
Раздел отчёта включает:
- Состояние (фенотип ClinVar), риск развития которого высок у пациента или его ближайших родственников, поскольку в образце были выявлены патогенные или вероятно патогенные варианты, ассоциированные с этим состоянием. В отчёте могут быть отмечены состояния, характерные для человека другого пола. Это означает, что обнаруженные варианты могут не влиять на риск развития этого состояния у пациента, но могут быть переданы им по наследству к человеку или могли быть унаследованы им от человека, который может находиться в зоне риска.
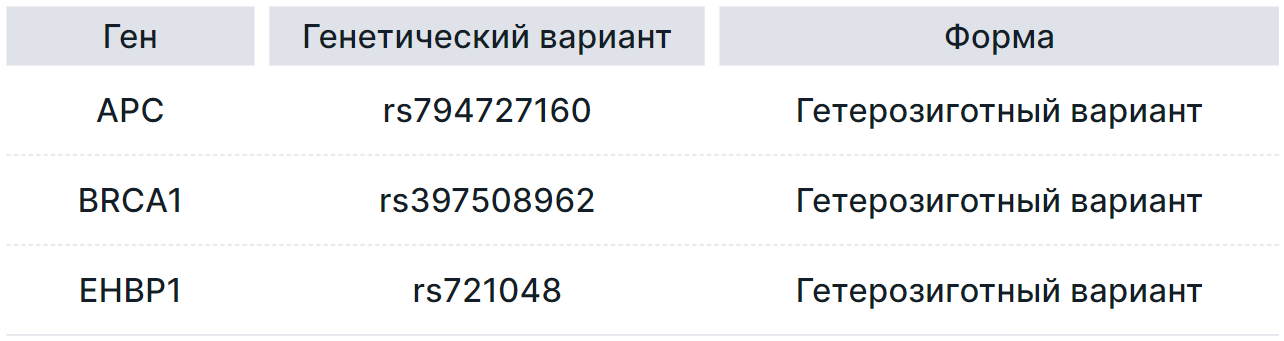
- Патогенные или вероятно патогенные варианты, ассоциированные с перечисленными выше состояниями. Занесены в таблицу со следующими колонками:
- Ген - название гена, в котором располагается вариант;
- Генетический вариант - идентификатор варианта в базе dbSNP (rsId);
- Форма, в которой был обнаружен вариант: гетерозиготная или гомозиготная.
- Общая рекомендация для пациента (консультация врача генетика).
- Информация для врача - краткая справка о том, как составляется отчёт.
2. Подробный отчёт#
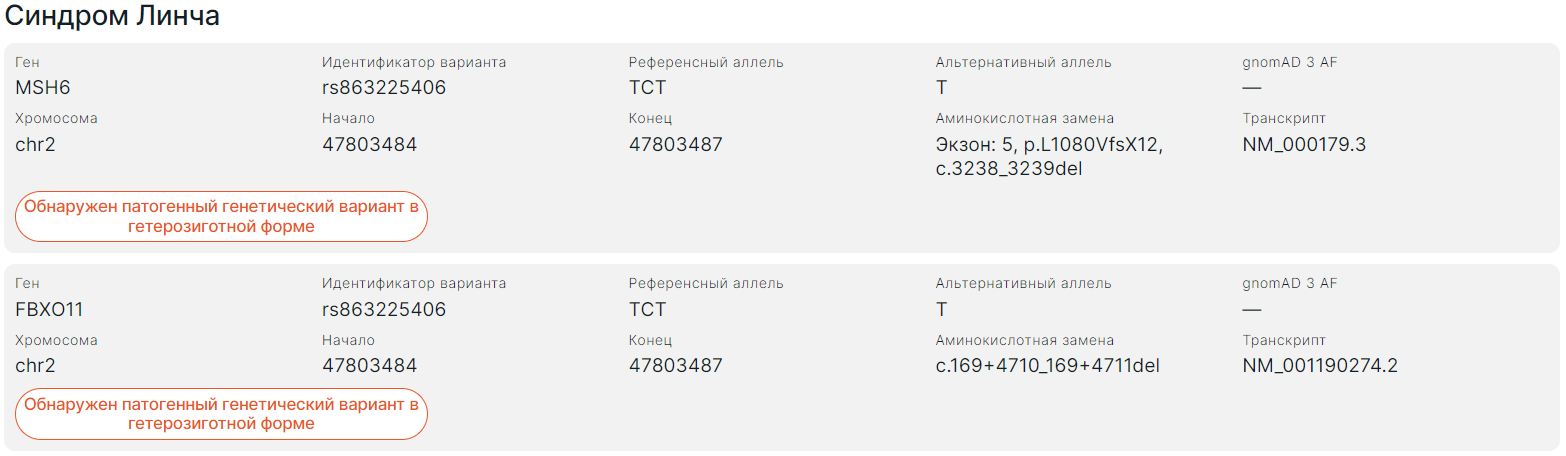
Подробный отчёт о состояниях, перечисленных в первом разделе. Для каждого состояния указано следующее:
- Название - соответствует полю "Имя в отчёте" из шаблона.
- Варианты, выявленные в образце, которые ассоциированы с данным состоянием (с указанными в шаблоне MedGen ID и/или именем в ClinVar). Кроме этого, каждый вариант соответствует следующим условиям:
- Патогенный или вероятно патогенный по данным ClinVar (поле "Clinical significance");
- Не гомозиготный по референсу (содержит хотя бы один альтернативный аллель);
- Суммарная частота аллеля в gnomAD 3 выше порога частоты крайне редких патогенных вариантов и ниже порога частоты слишком частых патогенных вариантов (пороги задаются в шаблоне).
- Рекомендации, которые могут помочь снизить риски возникновения данной патологии (добавляются в шаблоне).
3. Исключенные варианты#
Представляет варианты, которые были исключены из подробного отчёта о патологиях, по одному из следующих критериев (при этом всем перечисленным выше условиям для попадания в отчёт варианты удовлетворяют):
- Критерий 1: варианты, которые имеют слишком высокую частоту встречаемости (более 15%) среди представителей человеческой популяции (противоречит данным о возможной патогенности этих вариантов);
- Критерий 2: варианты, которые имеют генотип 1/1 и частоту встречаемости среди представителей человеческой популяции менее 1%. Согласно публикации, такие варианты рекомендуется исключать, поскольку они могли быть выявлены в результате ошибки генотипирования, проведенного с помощью технологии микрочипов.
- Критерий 3: варианты, которые имеют достоверность выявления ниже установленного порога достоверности
(порог задается в шаблоне).
Достоверность рассчитывается как (1 - Bin(n; m; p; 1)),
где Bin(n; m; p; 1) - это рассчитанная на основе биномиального распределения вероятность того, что вариант встретится ровно n раз среди m представителей (образцов) популяции;
n - количество представителей популяции (образцов), у которых был выявлен данный вариант;
m - общее количество представителей (образцов) в популяции;
p - вероятность встретить вариант среди представителей человеческой популяции (суммарная частота аллеля в базе данных gnomAD 3). Если данных о частоте в популяции нет, то вероятность принимается за 0,001.
Таким образом, из отчёта исключаются варианты, которые встретились более, чем n раз, среди m образцов популяции, но при этом имеют низкую частоту в человеческой популяции, т.е. информация о возможной патогенности таких вариантов противоречит слишком частой встречаемости среди представителей (образцов) популяции.
Значения n и m берутся из пользовательской базы частот (если она не была загружена в систему, фильтрация вариантов по критерию 3 происходить не будет).
Каждый вариант в отчёте представлен в следующем виде:#
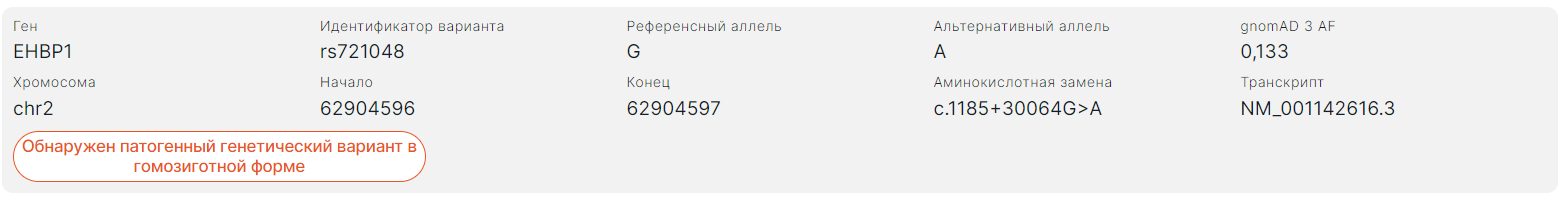
- Ген - название гена, в котором располагается вариант.
- Идентификатор варианта - идентификатор варианта из базы данных dbSNP.
- Референсный аллель варианта.
- Альтернативный аллель варианта.
- gnomAD 3 AF - суммарная частота аллеля в базе данных gnomAD (версия 3).
- Хромосома - хромосома, на которой находится вариант.
- Начало - начальная позиция варианта на хромосоме.
- Конец - конечная позиция варианта на хромосоме.
- Аминокислотная замена - номер экзона в транскрипте, который был затронут вариантом; аминокислотная и нуклеотидная замены в номенклатуре HGVS. Аминокислотная: префикс “p.” (protein) + референсная аминокислота + позиция аминокислоты в белке + новая аминокислота, получившаяся в результате замены. Нуклеотидная: префикс “c.” (coding; для замены в кодирующей последовательности) или “n.” (non-coding; для замены в некодирующей последовательности) + геномная позиция замещенного нуклеотида + референсный аллель > альтернативный аллель.
- Транскрипт - идентификатор основного транскрипта гена из базы RefSeq (NM_xxxxxx.x).
- Указание, в какой форме была обнаружена мутация: гетерозиготной или гомозиготной.
- Для исключенных из подробного отчёта вариантов - указание критерия, по которому они были исключены.
Экспорт отчёта#
Отчёт на основе фенотипа ClinVar можно скачать в формате PDF.
Для этого нажмите на кнопку  в
правом верхем углу страницы отчёта.
в
правом верхем углу страницы отчёта.