Биоинформатический отчёт
Вкладка "Биоинформатический отчёт" на странице образца включает основные метрики, которые помогут оценить качество исходных данных, качество выравнивания и покрытие данных, а также включает отчёты по выявленным SNVs/Indels.
На заметку
В биоинформатическом отчёте опухолевого образца из набора образцов опухоль/контроль представлена информация для файлов опухолевого и контрольного образцов:
В биоинформатическом отчёте образцов неопухолевой ткани и одиночных образцов опухоли представлена информация о файлах только этих образцов.
Биоинформатический отчёт может включать различные разделы в зависимости от формата загруженных данных и включенных в пайплайн стадий анализа. Возможные разделы отчёта:
- Статистика файла - у образцов, загруженных в формате FASTQ или BAM.
- Отчёт о качестве - у образцов, загруженных в формате FASTQ или BAM.
- Отчёт по выравниванию - у образцов, загруженных в формате FASTQ или BAM, для которых стадия "Выравнивание" была включена в пайплайн и завершилась успешно.
- Отчёт по покрытию - у образцов, загруженных в формате FASTQ или BAM, для которых стадия "Выравнивание" была включена в пайплайн и завершилась успешно.
- Отчёт по соматическим SNVs/Indels образца опухолевой ткани - у образцов, для которых стадии "Выявление соматических SNVs/Indels" и "Аннотация соматических SNVs/Indels" были включены в пайплайн и успешно завершились.
- Отчёт по герминальным SNVs/Indels - у образцов, для которых стадии "Выявление герминальных SNVs/Indels" и "Аннотация герминальных SNVs/Indels" были включены в пайплайн и успешно завершились.
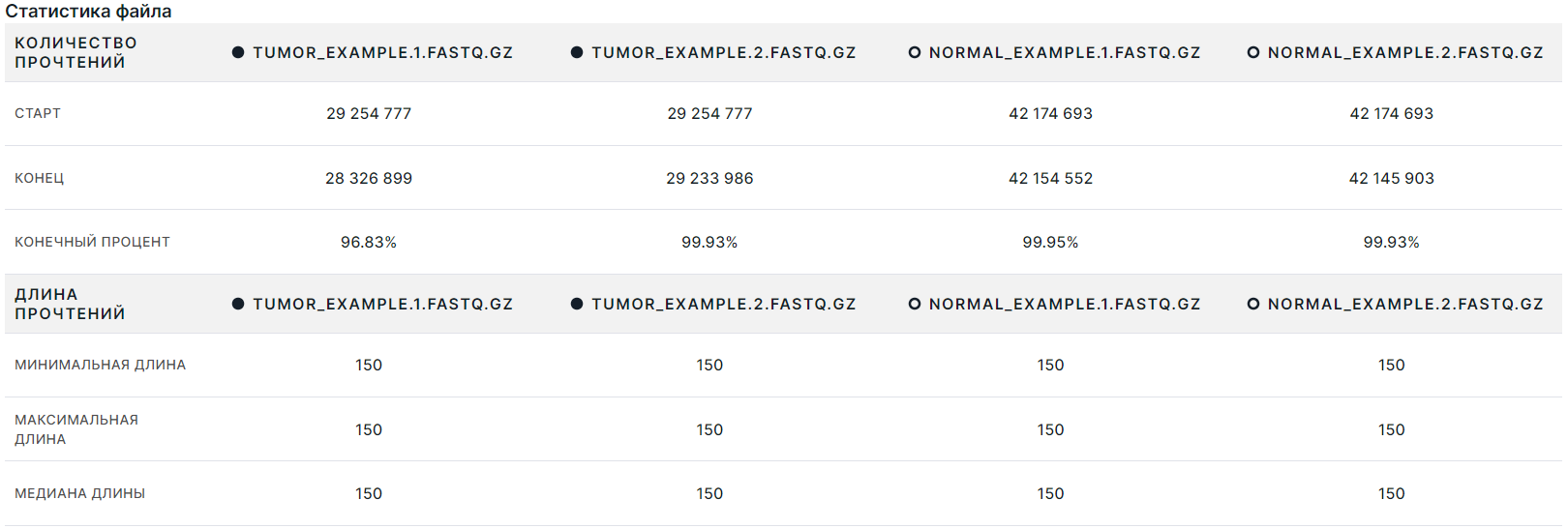
Статистика файла#
На заметку
Раздел есть только у образцов, загруженных в формате FASTQ или BAM.
- Данные по количеству прочтений в файле:
- Старт - количество прочтений в файле после его проверки, но до очистки.
- Конец - количество прочтений в файле, подсчитанных во время проверки качества после очистки.
- Конечный процент - доля прочтений (в процентах), которые остались в файле после очистки.
- Данные по длине прочтений в файле:
- Минимальная длина прочтения в файле образца.
- Максимальная длина прочтения в файле образца.
- Медиана длины - среднее значение длины прочтения в файле образца.
На заметку
Большое количество отфильтрованных во время очистки прочтений и значительное уменьшение длины прочтений говорит о низком качестве исходных данных. В таком случае рекомендуем рассмотреть возможность повторного секвенирования материала.
Отчёт о качестве#
На заметку
Раздел есть только у образцов, загруженных в формате FASTQ или BAM.
Отчёт содержит метрики финального (после очистки, если она проводилась) качества прочтений, рассчитываемые на стадии “Проверка качества и очистка”. Подробный отчёт по метрикам с визуализацией можно скачать на вкладке "Детали процесса анализа" в соответствующем разделе.
Метрики контроля качества прочтений#
| Метрика | Описание | Значение порога метрики, при котором прочтения в образце считаются качественными (значение по умолчанию, может быть изменено в настройках) |
| Total sequences | Количество прочтений | >200 000 |
| Length distribution | Распределение длин прочтений | Коротких прочтений (прочтений длиной ≤ 20 п.н.) менее 25% |
| Tiles sequence quality | Качество прочтений, поступивших из конкретных плиток проточных ячеек | Количество ячеек с плитками низкого качества с максимальным отклонением 1,165 менее 7 |
| First base sequence quality | Качество прочтения первых нуклеотидов последовательности | Худший нижний 10-й процентиль качества прочтения 3-х первых нуклеотидов больше 20 |
| Middle base sequence quality | Качество прочтения средних нуклеотидов последовательности | Худший нижний 10-й процентиль качества прочтения средних нуклеотидов больше 20 |
| Last base sequence quality | Качество прочтения конечных нуклеотидов последовательности | Худший нижний 10-й процентиль качества прочтения 3-х конечных нуклеотидов больше 20 |
| Overrepresented sequences | Перепредставленные последовательности - последовательности, которые составляют более 0,1% от общего числа последовательностей | Максимальный процент числа перепредставленных последовательностей в файле менее 1% |
| Adapter contaminated | Количество прочтений, содержащих адаптерные последовательности | Процент числа прочтений, контаминированных адаптерами, не более 1% |
| Base N content | Количество нераспознанных нуклеотидов N среди всех нуклеотидов последовательности | Максимальный процент количества N среди нуклеотидов не более 20% |
| GC content | Процентное содержание пары GC в прочтении | Наличие только одного пика содержания GC |
| Base sequence content | Процентное содержание четырех типов нуклеотидов в определенной позиции прочтения | Средняя разница AT менее 1%, максимальная разница AT менее 20%, средняя разница GC менее 1%, максимальная разница GC менее 20% |
Метрики, удовлетворяющие порогу качества,
отмечены ![]() , не удовлетворяющие -
, не удовлетворяющие - ![]() .
.
Отчёт по выравниванию#
На заметку
Раздел есть только у образцов, загруженных в формате FASTQ или BAM, для которых выравнивание было включено в пайплайн и успешно завершилось.
Раздел включает статистику выравнивания и метрики, описывающие качество выравнивания. При большом количестве ошибок секвенирования, неполных данных, высоком уровне контаминации или неверном определении источника (организма) образца метрики укажут на возможную проблему.
- Статистика выравнивания:
- Всего прочтений - суммарное количество прочтений в файле выравнивания.
- Картированные прочтения - количество прочтений образца, которые были картированы на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Некартированные прочтения - количество прочтений образца, которые не удалось картировать на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Прочтения с множественным картированием - количество прочтений, картированных на референсный геном несколько раз. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Прочтения с прямой цепи - количество прочтений с прямой цепи, картированных на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Прочтения с обратной цепи - количество прочтений с обратной цепи, картированных на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах). В нормальных данных количество прочтений с прямой и с обратной цепей одинаковое; разница в их количестве говорит о том, что в данных есть транслокации ДНК (например, инверсии).
- Парные прочтения - суммарное количество парных прочтений в файле выравнивания. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Картированные парные прочтения - количество парных прочтений образца, которые были картированы на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Правильно картированные парные прочтения - количество парных прочтений образца, правильно картированных на референсный геном. В скобках - доля количества таких прочтений от суммарного количества прочтений в файле (в процентах).
- Всего выравниваний - суммарное количество выравниваний прочтений образца на референсный геном.
- Выравнивания с множественным картированием - количество выравниваний одного прочтений на референсный геном несколько раз. В скобках - доля количества таких выравниваний от суммарного количества выравниваний в файле (в процентах).
- Выравнивания с прямой цепи - количество выравниваний прочтений с прямой цепи на референсный геном. В скобках - доля количества таких выравниваний от суммарного количества выравниваний в файле (в процентах).
- Выравнивания с обратной цепи - количество выравниваний прочтений с обратной цепи на референсный геном. В скобках - доля количества таких выравниваний от суммарного количества выравниваний в файле (в процентах).
- Percent duplication - доля выровненной последовательности, которая была помечена как дубликат, (в процентах). Статистика включена в отчёт, если у образца была включена в пайплайн и успешно завершилась стадия "Пометка дубликатов".
- Метрики качества выравнивания:
| Метрика | Описание метрики | Значение порога метрики по умолчанию (может быть изменено в настройках) |
| Mapped reads | Доля количества картированных прочтений (в процентах) от суммарного количества прочтений в файле выравнивания. | ≥ 85 |
| Multiple alignments | Доля множественных выравниваний одного и тоже прочтения на геном (в процентах) от суммарного количества выравниваний в файле выравнивания. | ≤ 15 |
| Forward/reverse balance | Разница в количестве прочтений с прямой цепи и прочтений с обратной цепи от суммарного количества прочтений в файле выравнивания (в процентах). | ≤ 10 |
| Paired mapped reads | Доля количества картированных парных прочтений (в процентах) от суммарного количества прочтений в файле выравнивания. Есть только для парных образцов секвенирования. | ≥ 80 |
| Paired properly mapped reads | Доля количества правильно картированных парных прочтений (в процентах) от суммарного количества прочтений в файле выравнивания. Есть только для парных образцов секвенирования. | ≥ 75 |
Метрики, удовлетворяющие порогу качества,
отмечены ![]() , не удовлетворяющие -
, не удовлетворяющие - ![]() .
.
Отчёт по покрытию#
На заметку
Раздел есть только у образцов, загруженных в формате FASTQ или BAM, для которых выравнивание было включено в пайплайн и успешно завершилось.
Отчёт по покрытию генома данными позволяет оценить, насколько информативен анализ этих данных в целом.
- Coverage per nucleotide - понуклеотидное покрытие на геноме. Порог метрики - ≥ 0.1. Если метрика
удовлетворяет порогу качества, она отмечена
 , а если не удовлетворяет, то
, а если не удовлетворяет, то  .
. - PCT selected bases - доля (в процентах) PF_BASES_ALIGNED (количество уникальных оснований, прошедших фильтрацию, которые выровнены на референсный геном с оценкой картирования > 0), расположенных в или около района "приманки" (baited region), который вычисляется по формуле (ON_BAIT_BASES + NEAR_BAIT_BASES)/PF_BASES_ALIGNED, где ON_BAIT_BASES - количество PF_BASES_ALIGNED, картированных на район "приманки" генома; NEAR_BAIT_BASES - количество PF_BASES_ALIGNED, картированных на область фиксированного интервала, содержащего район "приманки", но не на сам этот район. Метрика включена в отчёт для образцов, являющихся результатом секвенирования с таргетной панелью, если успешно завершилась стадия "Вычисление покрытия".
- PCT usable bases on target - доля (в процентах) количества выровненных, дедублированных, целевых оснований из всех доступных оснований, прошедших фильтрацию. Метрика включена в отчёт для образцов, являющихся результатом секвенирования с таргетной панелью, если успешно завершилась стадия "Вычисление покрытия".
- Mean target coverage - среднее покрытие целевой области. Метрика включена в отчёт для образцов, являющихся результатом секвенирования с таргетной панелью, если успешно завершилась стадия "Вычисление покрытия".
- Median target coverage - серединное (медиана) покрытие целевой области. Метрика включена в отчёт для образцов, являющихся результатом секвенирования с таргетной панелью, если успешно завершилась стадия "Вычисление покрытия".
Отчёт по соматическим или герминальным SNVs/Indels#
На заметку
Раздел "Отчёт по соматическим SNVs/Indels образца опухолевой ткани" есть у тех образцов, для которых
стадия "Выявление соматических SNVs/Indels"
была включена в пайплайн, а стадия "Аннотация соматических SNVs/Indels" завершилась успешно.
Раздел
"Отчёт по герминальным SNVs/Indels образца опухолевой ткани"
есть у тех одиночных образцов опухолевой ткани (анализируемых без контроля), для которых
стадия "Выявление герминальных SNVs/Indels"
была включена в пайплайн, а стадия "Аннотация герминальных SNVs/Indels" завершилась успешно.
Раздел
"Отчёт по герминальным SNVs/Indels образца неопухолевой ткани"
есть у образцов опухолевой ткани (анализируемых с контролем) и у образцов неопухолевой ткани, для которых
стадия "Выявление герминальных SNVs/Indels"
была включена в пайплайн, а стадия "Аннотация герминальных SNVs/Indels" завершилась успешно.
- Количество вариантов:
- Всего - суммарное количество однонуклеотидных вариантов (single-nucleotide variants; SNVs) и коротких инсерций/делеций (indels), выявленных в образце. В скобках указано количество генов, в которых расположены выявленные варианты.
- Количество SNV - количество однонуклеотидных вариантов, выявленных в образце.
- Количество INDEL - количество коротких инсерций/делеций, выявленных в образце.
- Позиция в геноме:
- Exonic - количество вариантов, расположенных в экзоне:
- Frameshift - количество инсерций или делеций, вызывающих сдвиг рамки считывания;
- Start loss - количество вариантов, вызывающих мутацию старт-кодона в non-start кодон;
- Stop gain - количество вариантов, приводящих к появлению стоп-кодона;
- Stop loss - количество вариантов, вызывающих мутацию стоп-кодона в non-stop кодон;
- Missense - количество вариантов, приводящих к появлению кодона, который кодирует другую аминокислоту;
- Inframe indel - количество инсерций или делеций одного или нескольких кодонов;
- Synonymous - количество вариантов, приводящих к появлению кодона, который кодирует ту же аминокислоту;
- 5'UTR - количество вариантов, попадающих в 5′-нетранслируемую область;
- 3'UTR - количество вариантов, попадающих в 3′-нетранслируемую область.
- Intronic - количество вариантов, расположенных в интроне.
- Intergenic - количество вариантов, расположенных в межгенной области:
- Upstream - количество вариантов, расположенных перед геном;
- Downstream - количество вариантов, расположенных после гена.
- In splice site - количество вариантов, расположенных в сайте сплайсинга.
- In non-protein-coding transcript - количество вариантов, расположенных в некодирующем транскрипте.
- Известные варианты - варианты, информация о которых содержится в различных базах данных, таких как dbSNP, COSMIC (если база была загружена в виде пользовательской аннотации), 1000 Genomes, gnomAD 3, ClinVar (с указанием количества вариантов с определённой клинической значимостью фенотипа; определение значений можно посмотреть тут), dbNSFP.
Экспорт отчёта#
Биоинформатический отчёт можно скачать в формате PDF.
Для этого нажмите на кнопку  в
правом верхем углу страницы отчёта.
в
правом верхем углу страницы отчёта.