Genomic predictions
After the "Unify SNVs/Indels" task has been successfully completed, the genomic predictions pipeline can be launched for non-tumor samples if at least one of the following tasks was included in the analysis workflow: "Oligogenic risk scores calculation", "Polygenic risk scores calculation", "Pharmacogenetic calculation", or "Ancestry analysis". Please note that to include genomic predictions in the workflow of a sample uploaded in VCF or GT format, you need to select the setting preset that includes one of the above parameters during the sample set composing.
If any of the following tasks fail, the sample analysis stops. However, if quality control and imputation are included in the analysis workflow, and if the sample does not meet the quality control criteria, the "Genomic predictions" stage interrupts, but the sample analysis can proceed with report generation.
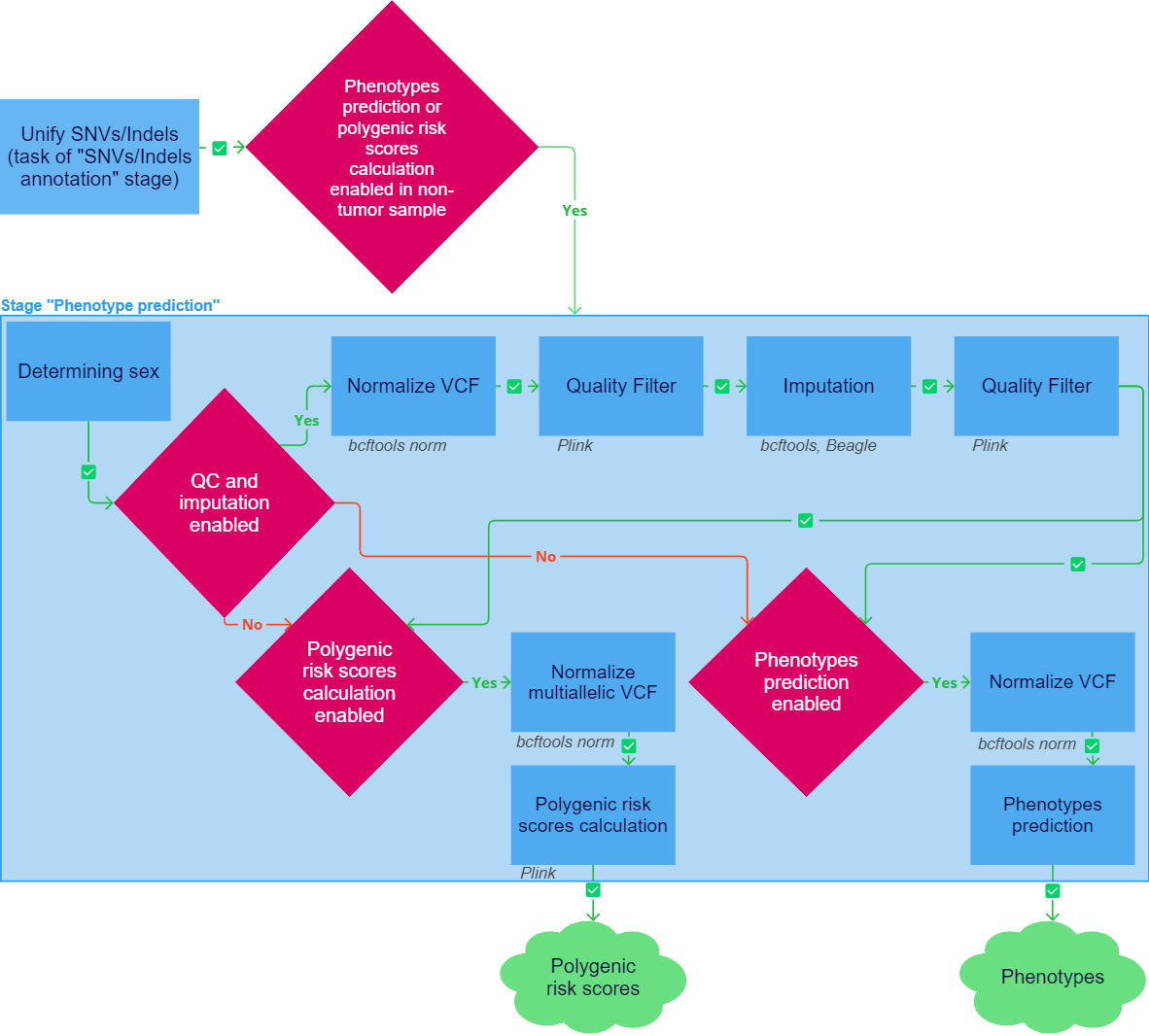
The "Genomic predictions" stage may include the following tasks:
- Determining sex: imputation from the X-chromosome homozygosity level data. The X-chromosome inbreeding coefficient F is calculated using the following formula:
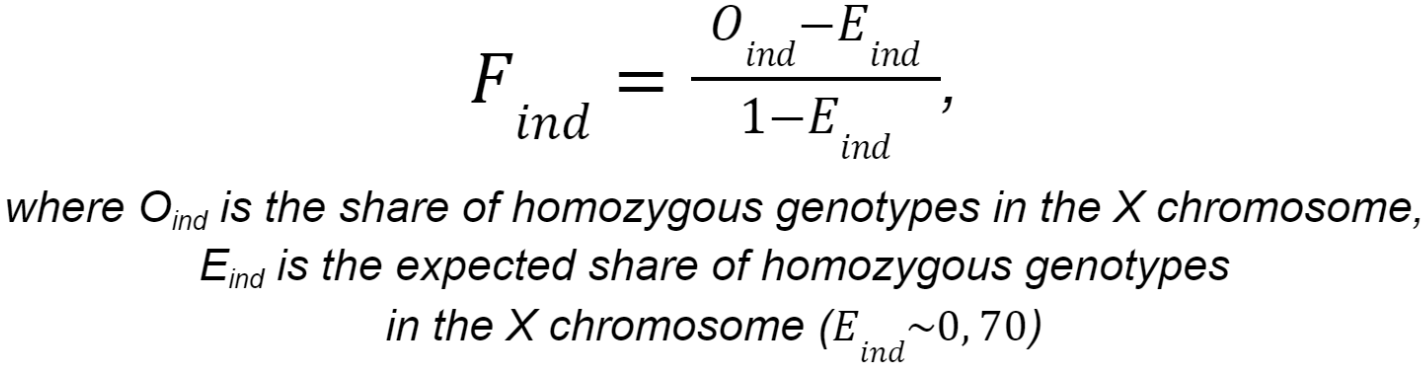
If F < 0.2, then the sex is determined as female. If F > 0.8, then the sex is determined as male. If 0.2 < F < 0.8, then the sex cannot be determined unambiguously.
The patient's sex is used to generate the report on polygenic traits. If the user manually entered sex, this value is used; otherwise, the sex determined from genetic data is used.
Quality control and imputation: if the "Enable QC and imputation" parameter is turned on.
2.1. Normalize VCF:
bcftools norm checks if reference alleles in the file match the reference; splits multiallelic sites into biallelic records; outputs only the first instance if a record is present multiple times.
Compressing a file into a GZIP archive using bgzip. The resulting file can be downloaded in the "Result files" section in the "Normalize VCF" task details ("Download VCF_GZ").
Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download VCF_TBI").
2.2. Quality Filter:
bcftools filter excludes sites for which genotype information is missing for the site (e.g., ./.).
PLINK converts a VCF file into a PLINK 2 binary fileset (i.e. a way to represent genotype calls), and removes all but one instance of each duplicate-ID variant (ignoring the missing ID). A file with a detailed description of this task log can be downloaded in the "Result files" section in the "Quality Filter" task details ("Download convert log TXT").
Quality control. The checks are performed throughout samples and single nucleotide polymorphisms (SNPs), those not meeting the criteria are removed. All of the quality control procedures are conducted through PLINK. QC process includes pre-stage (sample and SNP call rate, and sex check) and iterative stages that are repeated iteratively until errors are no longer detected.
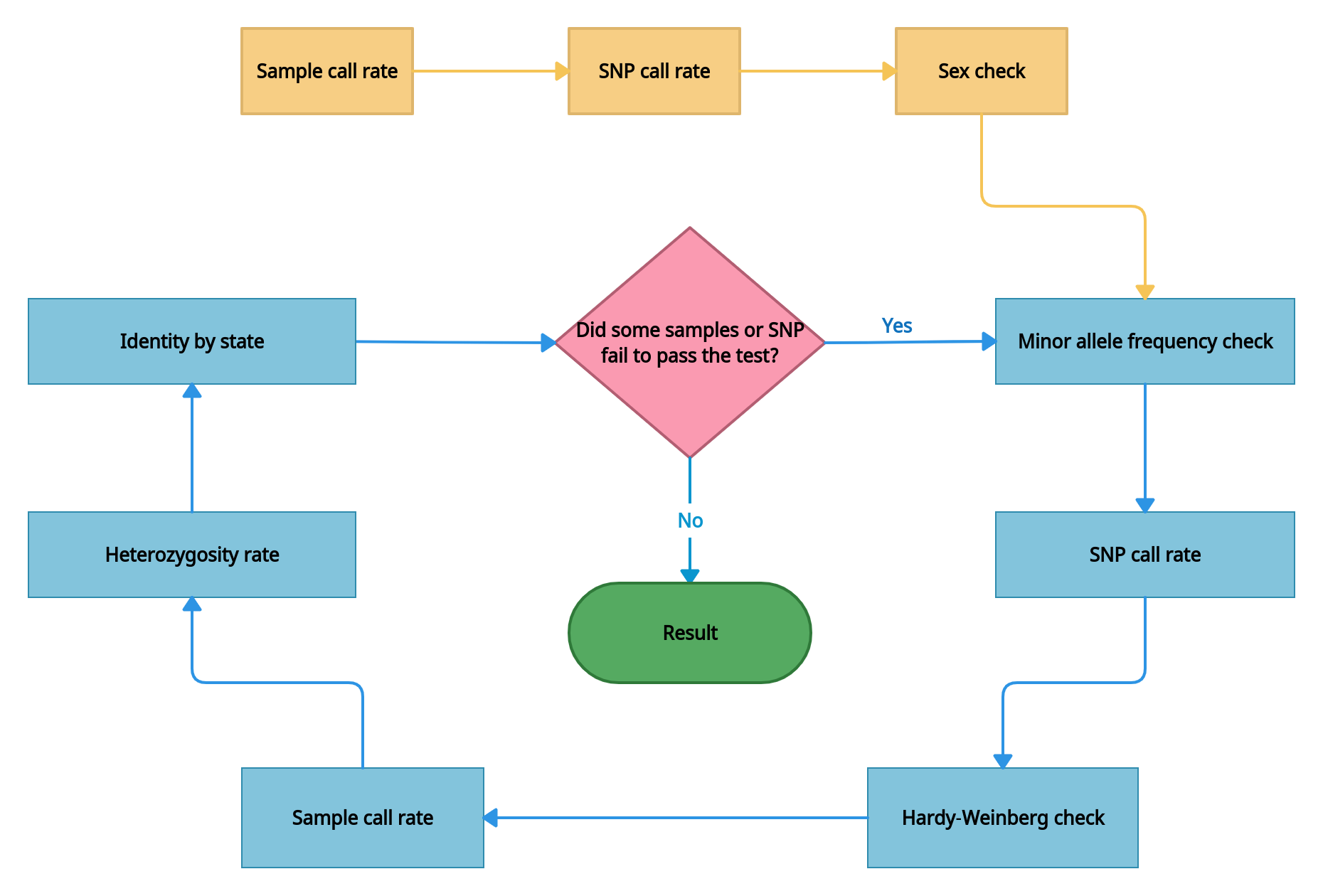
- Checking the data for samples with extremely low call rate. Samples with high missing call rate are implications of poor DNA quality. They are removed from analysis. The sample call rate threshold is > 0.5.
- Checking the data for SNPs with extremely low call rate. The missing call rate of SNP is the proportion of samples whose genotypes are not called for a given SNP. SNPs with a high missing genotype rate (generally, >5%) imply some problems with the genotyping process, so these SNPs are eliminated from analysis. The SNP call rate threshold is > 0.5.
- Sex check and removing samples with wrong sex assigned. Sex check is based on estimation of X-linked SNPs heterozygosity. By default, X-chromosome inbreeding coefficient F estimates smaller than 0.2 yield female samples, and values larger than 0.8 yield male samples.
- Minor allele frequency (MAF) check. SNPs with MAF less than 1% are excluded from subsequent analysis as current SNP-chips genotyping rare variants (i.e. a locus with MAF< 1%) is difficult and error-prone. Thus, very low-frequency alleles are likely to represent genotyping error and may result in spurious associations.
- SNP filtration by call rate with a threshold > 0.98.
- Hardy-Weinberg equilibrium (HWE) check. Under Hardy-Weinberg’s assumptions, allele and genotype frequencies can be estimated from one generation to the next. It is noted that departure from Hardy-Weinberg equilibrium can occur due to selection, population admixture, cryptic relatedness, genotyping error, and genuine genetic association. So it is beneficial to check whether the SNPs deviate from HWE for quality control.
- Sample filtration by call rate with a threshold > 0.98.
- Sample heterozygosity. The proportion of heterozygous genotypes across an sample's genome can detect several issues with genotyping, like sample contamination, inbreeding. Samples that deviate ± 3 SD (standard deviation) from the sample's mean heterozygosity are removed from the analysis.
- Identity by state check. A high degree of relatedness between samples can also generate inflation of the
association. To investigate the cryptic relatedness we calculate kinship matrix and filter samples with close
relationships. Kinship coefficient is > 0.0925.
A file with a detailed description of the quality control task log can be downloaded in the "Result files" section in the "Quality Filter" task details ("Download QC log TXT").
Second check and filtering of variants and generation of a new PLINK 2 binary fileset set with filtered samples and SNPs using PLINK.
A file with a detailed description of this task log can be downloaded in the "Result files" section in the "Quality Filter" task details ("Download merge log TXT").
A file with SNPs removed from the analysis can be downloaded in the same section ("Download Skipped variants TXT"). For each SNP, the reason for removing and the QC iteration run number at which the removing occurred are indicated.
The full data quality control report can be opened in the same section ("Open QC report HTML").
A file with samples removed from the analysis can be downloaded in the same section ("Download Removed samples TXT"). For each sample, the reason for removing and the QC iteration run number at which the removing occurred are indicated.Excluding SNPs that did not pass filtering from analysis using vcftools.
Compressing a file into a GZIP archive using bgzip. The resulting file can be downloaded in the "Result files" section in the "Quality Filter" task details ("Download Filtered VCF_GZ").
Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download Filtered VCF_TBI").
2.3. Imputation is a statistical method for reconstructing missing genetic data based on haplotype analysis in a reference set.
bcftools index creates an index for a VCF file.
Dividing variants by chromosomes for parallel imputation using bcftools view.
Beagle phases genotypes and imputes ungenotyped markers.
Concatenating the imputed variants, divided by chromosomes, into a single VCF file using bcftools concat.
bcftools index creates an index for a VCF file.
Comparison of the original file and the file obtained after imputation, and generating a file with unimputed variants using vcftools.
Compressing a file with unimputed variants into a GZIP archive using bgzip.
bcftools index creates an index for a file with unimputed variants.
bcftools filter includes only imputed sites for which DR2 (dosage R-squared) > 0.3.
bcftools index creates an index for a file with imputed and filtered variants.
Concatenating the unimputed variants and filtered imputed variants into a single VCF file using bcftools concat.
bcftools index creates an index for a merged file.
A file with a detailed description of the imputation task log can be downloaded in the "Result files" section in the "Imputation" task details ("Download Impute log TXT").
A file with unimputed and filtered imputed variants in VCF format can be downloaded in the same section ("Download Imputed VCF_GZ").
An index file for VCF file can be downloaded in the same section ("Download Imputed VCF_TBI").2.4. Quality Filter: the task described above is repeated.
info
If a sample fails a quality control check, the "Genomic predictions" stage is interrupted and no oligogenic/polygenic risk scores calculation and/or ancestral analysis occurs. Sample analysis can proceed with report generation. You can upload the sample in VCF or GT format again with the setting preset with QC and imputation disabled, but the results may be insufficient in this case. Alternatively, you can upload the sample in FASTQ or BAM format. This may also solve the problem.
Polygenic risk scores calculation if the "Run polygenic risk scores calculation" parameter is enabled:
3.1. Normalize multiallelic VCF:
bcftools norm left-aligns and normalizes indels; checks if reference alleles in the file match the reference; joins biallelic sites into multiallelic records.
Compressing a file into a GZIP archive using bgzip. The resulting file with normalized variants can be downloaded in the "Result files" section in the "Normalize multiallelic VCF" task details ("Download VCF_GZ").
Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download VCF_TBI").
3.2. Polygenic risk scores calculation: applying a linear scoring to each sample using PLINK. Variants for which genotype information is missing for the site (genotype ./. etc.), variants without ID, or with mismatched allele codes are not taken into account in the analysis. The patient's genetic data must include the variants represented in the polygenic risk model, with the exception of a small proportion set by the threshold value. Polygenic risk scores calculation is considered possible if the user's genetic data contains at least 95% of all variants represented in the model.
The polygenic risk score (PRS) represents the total number of genetic variants (SNPs) that an individual has to assess their heritable risk of developing a particular phenotype. For each trait, the polygenic risk score is calculated using the following formula:
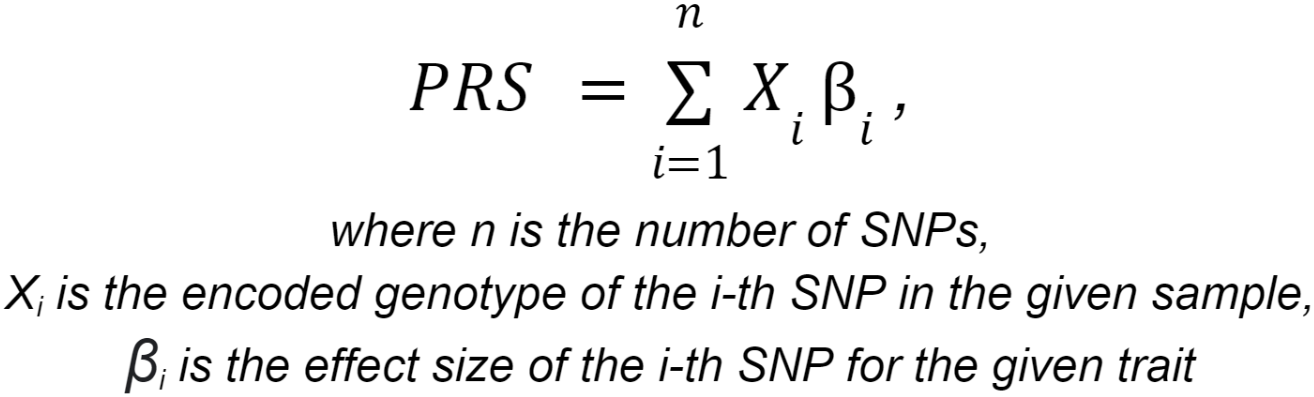
Genotypes are coded as follows: considering allele A as the effect allele and allele G as the ineffect allele,
the numerical code for genotype AA is 2, genotype AG is 1, and genotype GG is 0.
The estimated effect sizes of SNPs are calculated based on genome-wide association study (GWAS) data, which
match phenotypic traits with genomic variants in human populations.
The polygenic risk score reflects an individual's estimated genetic predisposition to the given trait and can
be used as a predictor of that trait in a predictive model. In other words, the PRS estimates how likely an
individual is to have the given trait based only on genetic data and without taking into account environmental factors.
The polygenic risk scores are calculated for the following traits:
- Height
- Weight
- Body Mass Index (BMI)
- Predisposition to being overweight
- Predisposition to Prostate Cancer
- Predisposition to Breast Cancer
- Predisposition to Coronary Artery Disease
- Predisposition to Inflammatory Bowel Disease
- Predisposition to Type 2 diabetes
- Predisposition to Colorectal Cancer
For each trait, three files are generated as a result, which can be downloaded in the "Result files" section in the "Polygenic risk scores calculation" task details:
- A file with a detailed description of PRS calculation log ("Download [the trait name] Prs log TXT").
- A file with the summary risk score for this sample ("Download [the trait name] Score TSV"). You can also open it in Google Sheets.
- A file with a list of variant IDs used to calculate PRS ("Download [the trait name] Used Variants TSV"). You can also open it in Google Sheets.
Calculation of oligogenic risk scores if the "Run oligogenic risk scores calculation" parameter is enabled:
4.1. Normalize VCF:
bcftools norm left-aligns and normalizes indels; checks if reference alleles in the file match the reference; splits multiallelic sites into biallelic records; outputs only the first instance if a record is present multiple times.
Compressing a file into a GZIP archive using bgzip. The resulting file with normalized variants can be downloaded in the "Result files" section in the "Normalize VCF" task details ("Download VCF_GZ").
Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download VCF_TBI").
4.2. Calculation of oligogenic risk scores: the prediction of oligogenic traits is based on single nucleotide polymorphisms (SNPs) associated with specific trait manifestations, using models built on multinomial logistic regression (MLR).
Calculated oligogenic risks:
- Hair color;
- Eyes color;
- Skin color;
- Freckling;
- Lactose intolerance;
- Bitter taste;
- Blood type;
- Alcohol metabolism;
- Ear wax;
- Body odour;
- Misophonia;
- Sleep movement risk;
- Photic sneeze reflex sensitivity.
Each trait and its corresponding prediction model are described in detail in the section on the report on oligogenic traits.
The resulting file with predicted oligogenic traits can be downloaded in the "Result files" section in the "Calculation of oligogenic risk scores" task details ("Download Classifier results JSON").
After the "Genomic predictions" stage, the analysis can proceed with report generation.