ACMG Secondary Findings Report
The report includes the variants discovered in the sample, which are located in the genes from the Secondary Findings (SF) list of American College of Medical Genetics and Genomics (ACMG). For these genes, specific mutations are known to be causative of disorders with defined phenotypes that are clinically actionable by an accepted intervention. The ACMG recommends that pathogenic or likely pathogenic variants detected in any of these genes should be reported as they are of medical relevance and could be used in the future to inform clinical treatment.
caution
Conditions for the report generation#
The report is generated for the sample if the following conditions are met:
- The sample is uploaded in FASTQ or BAM format or as a VCF file with a single sample.
- The sample analysis was successfully completed (that is, all stages of the analysis have the status "Complete").
- For a sample, the variant annotation stage was successfully completed: "Germline SNVs/Indels annotation" stage if Germline origin was selected when creating a report template, or "Somatic SNVs/Indels annotation", if Somatic origin.
- The report template including "SNVs/Indels in ACMG SF genes" block was created before the sample was processed.
- The report template including "SNVs/Indels in ACMG SF genes" block is active.
Report content#
The report includes two sections with lists of variants discovered in the sample, which are filled in according to the following scheme:
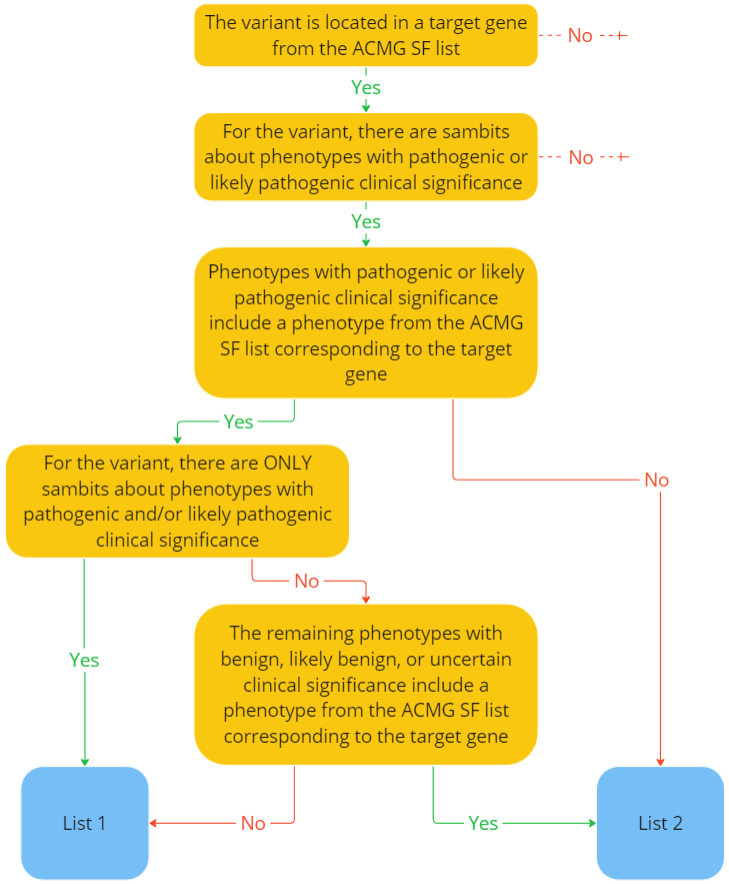
TIER I: The most important variants for interpretation for which the following conditions are met:
- The variant is located in a gene from the ACMG SF list;
- The variant is associated according to ClinVar with a phenotype only of pathogenic or likely pathogenic clinical significance that is listed in the ACMG SF list for the gene in which the variant is located;
- The allele frequency in gnomAD 3 is empty or below the frequency threshold for too frequent pathogenic variants, specified in the report template block "SNVs/Indels in ACMG SF genes" (recommended default value = 0.03);
- The variant site has at least one alternative allele (i.e. has the genotype 1/1, 1|1, 0/1, 1/0, 0|1, etc.) in the case of a VCF file with a single sample;
- The variant meets all the conditions specified in the ACMG SF list
for the gene in which the variant is located. These conditions may be associated with:
- the clinical significance of the phenotype (as noted above, the report primarily includes variants associated with a phenotype only with pathogenic or likely pathogenic clinical significance according to ClinVar; this condition is not required only for variants in HFE gene);
- type of inheritance;
- the number of variants discovered in the sample and suitable for the conditions that are located in one gene from the ACMG SF list (for example, for some variants with an autosomal recessive inheritance to be included in the report, at least two such variants must be discovered in the sample);
- the variant origin (Tier 1 includes only those variants for which there is no “somatic” among the allele origins);
- the genotype (for example, some variants are reported only if they are homozygous in alternative allele);
- specific nucleotide and amino acid substitution of the variant.
TIER II: Variants that require further analysis for which the following conditions are met:
- The variant is located in a gene from the ACMG SF list;
- The variant is associated with at least one phenotype of pathogenic and/or likely pathogenic clinical significance according to ClinVar;
- The allele frequency in gnomAD 3 is empty or below the frequency threshold for too frequent pathogenic variants, specified in the report template block "SNVs/Indels in ACMG SF genes" (recommended default value = 0.03);
- The variant site has at least one alternative allele (i.e. has the genotype 1/1, 1|1, 0/1, 1/0, 0|1, etc.) in the case of a VCF file with a single sample;
- The variant does not meet at least one of the conditions in Tier I:
- the variant is not associated according to ClinVar with a phenotype that is listed in the ACMG SF list for the gene in which the variant is located;
- according to ClinVar, the variant is associated with at least one phenotype of benign, likely benign, or uncertain clinical significance, which is listed in the ACMG SF list for the gene in which the variant is located;
- the variant does not meet at least one of the conditions specified in the ACMG SF list for the gene in which the variant is located.
note
There is an additional condition for inclusion of variants in the TTN gene to the ACMF SF report: only variants with a high impact of consequences on genes are included to the report: splice acceptor (variant hitting a splice acceptor site), splice donor (variant hitting a splice donor site), stop gained (variant causing a STOP codon) or frameshift (insertion or deletion causing a frame shift).
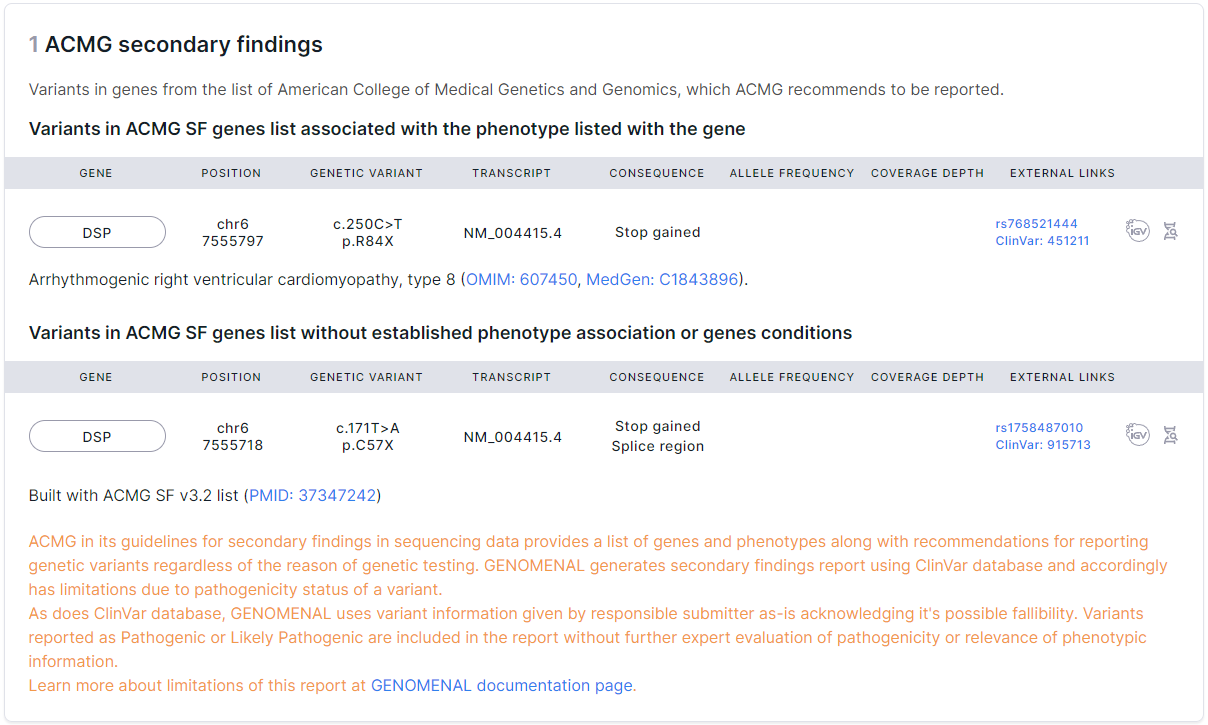
Each section contains a table of variants with the following columns:
- Gene is the common name of the gene in which the variant is located. If you click on a gene, you will see a window with all the gene transcripts:
![]()
You can find the description of the transcripts' table columns here.
- Position is a coordinate of the variant in the genome (chromosome + start position).
- Genetic variant is the nucleotide and amino acid substitutions using the HGVS notation. Nucleotide substitution: “c.” (coding; for a substitution in the coding sequence) or “n.” (non-coding; for a substitution in the non-coding sequence) prefix + genomic position of the substituted nucleotide + reference allele > alternative allele. Amino acid substitution: “p.” prefix (protein) + reference amino acid + amino acid position in protein + new amino acid resulting from the substitution.
- Transcript is the main transcript ID from the RefSeq database (NM_xxxxxx.x). If you click on a transcript, you will see the same window with all the gene transcripts, as when you click on the "Gene" field.
- Consequence is the effect of the variant on genes. A detailed description of the possible values can be found here.
- Allele frequency is an alternative allele frequency for the sample (in percentages).
- Coverage depth is a sequencing depth; the total number of reads of the sequence overlapping the variant position for the sample.
- External links are links to pages with variant information in dbSNP, ClinVar and COSMIC (if it was uploaded as a custom annotation).
- Links to the variant in embedded services:
 is a module for visualization of variant on the genome,
is a module for visualization of variant on the genome,  is
a variant details page in SNV Viewer
("Annotation" tab).
is
a variant details page in SNV Viewer
("Annotation" tab).
Below the variant row, there can be phenotypes that the variant is associated with according to ClinVar (with pathogenic or likely pathogenic clinical significance) and that are listed in the ACMG SF list for the gene in which the variant is located. Links to OMIM and/or MedGen are provided for each phenotype.
Next, there can be the variant interpretation text, if it was added as described here.
At the end of the report, there is the version of the ACMG SF list used to generate the report.
Potential findings#
Variants included in the ACMG SF report can be used as potential findings. To do this, you need to turn on the corresponding toggle in the report template. Potential findings can include Tier I variants (“Use variants in ACMG SF genes list associated with the phenotype results as potential findings” option) and/or Tier II variants (“Use variants in ACMG SF genes list without established phenotype association or genes conditions as potential findings" option).
Report limitations in ClinVar as a database source#
ACMG secondary findings report generation has limitations of phenotypic and pathogenicity data due to variant annotation using ClinVar database.
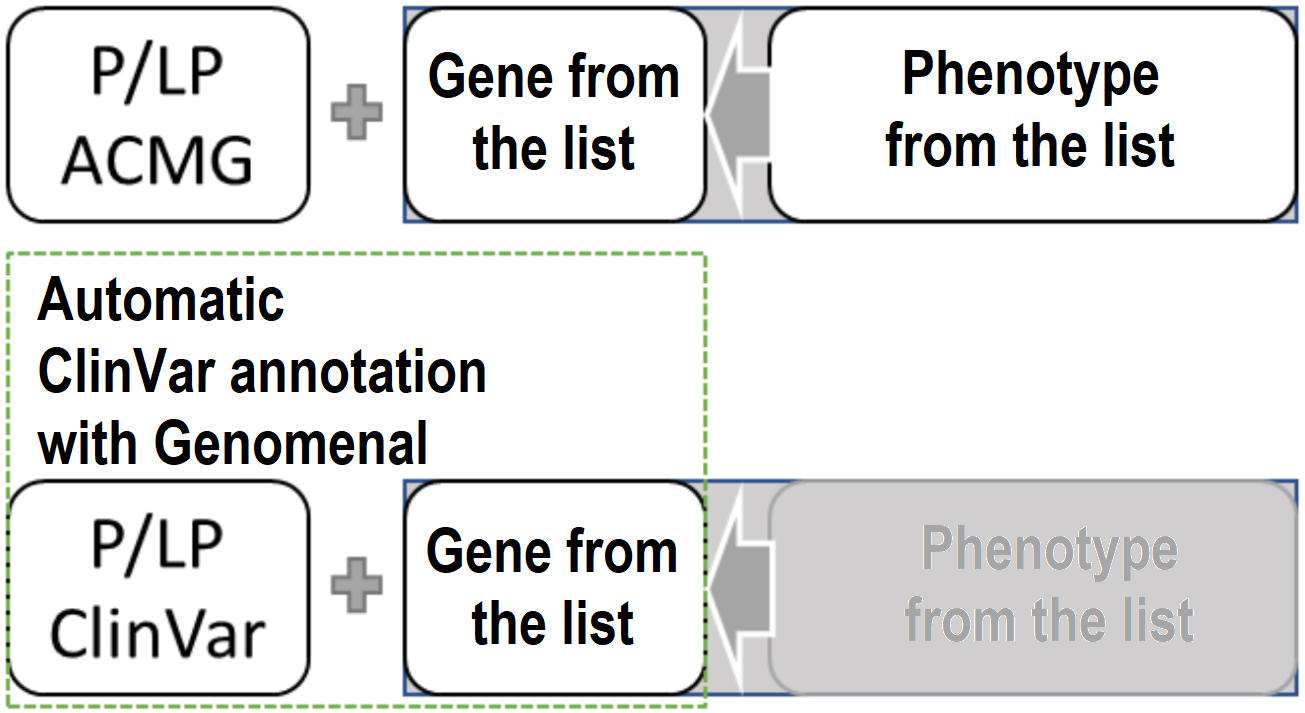
ACMG guidelines for secondary findings in sequencing data provide a list of genes and corresponding phenotypes along with recommendations for reporting genetic variants regardless of the reason of genetic testing. The following three conditions should be satisfied:
- The variant affects a gene from the list of secondary findings;
- The variant is reported as associated with the phenotype (condition, disease) listed with the gene in the list for secondary findings, or there is a possibility to establish such an association.
- The variant must meet all additional conditions specified in the list of secondary findings, which often include pathogenic or likely pathogenic clinical significance of the phenotype.
Briefly, it can be described as:
Limitations of phenotypic data#
For some of variants in ClinVar, it is impossible to obtain information about phenotype (or condition, or disease). Missense of phenotypic information in Clinvar can stem from inconsistencies in "Conditions" field in reports submitted by laboratories. The field can contain values "not reported", "not specified", or can be filled with all the diseases associated with any of the pathogenic variants in the gene. In the latter case, correct conclusions can be only by a specialist. Moreover, mutations in different portions of the gene can result in different genetic diseases, while the list for secondary findings can contain only a fraction of these phenotypes. Considering these and other factors, the ACMG Secondary Findings Report by Genomenal includes two lists of variants: those for which the phenotypes in the ACMG SF list match the condition reported for the variants in ClinVar, and those that are located in a gene in the list but have a discordant phenotype.
Therefore, the interpretation of the variants in the second section of the report must be approached with caution.Limitations of pathogenicity data#
Usually variants are classified according to ACMG guidelines. ClinVar database is populated by reports of pathogenicity from laboratories assuming that the responsible submitter fully described submitted variants according to ACMG recommendations. Similarly, Genomenal follows the same assumptions and variants are included in the report if they are reported as Pathogenic or Likely Pathogenic without further expert evaluation of pathogenicity.
Additionally, new evidence might emerge in literature about functional effect or lack of it for a specific variant. It can result in a revision of variant pathogenicity but will not be instantly reflected in ClinVar. Undoubtedly, systematical review of variants' pathogenicity status is labour-consuming and can lag compared to emergency of new scientific evidence. These factors can result in overreporting of variants, or presence of outdated data, or underreporting of variants for which new data has emerged but it is not reflected in ClinVar. Some variants can be omitted if they pathogenicity is yet unknown and not detected by anyone.
Thus, the report can include the following variants:
- with imprecise or erroneous pathogenicity status reported by the submitting laboratory;
- with pathogenicity revised due to newly emerged data (confirmed pathogenic by new data).
The following variants will not be included in the report:
- not yet reported in ClinVar or newly discovered;
- with pathogenicity revised due to newly emerged data (confirmed benign by new data).
caution
By using ACMG Secondary Findings report, you agree that you understand above mentioned limitations, and if clarification is needed, you are willing to discuss the result with medical specialist.
Report export#
ACMG Secondary Findings report can be downloaded in PDF format. To do this, click on
the button  in the upper right corner of the
report page.
in the upper right corner of the
report page.