Annotation
After successful upload, identification and verification, the sample analysis continues with the main stage of the analysis, such as annotation. If the uploaded annotation contains SNVs/Indels, the "Germline SNVs/Indels annotation" stage (if the sample was uploaded as a non-tumor sample) or "Somatic SNVs/Indels annotation" stage (if the sample was uploaded as a tumor sample) is launched. If SNVs/Indels are not found in the uploaded annotation, but there are structural variations, the "Germline SV annotation" stage (if the sample was uploaded as a non-tumor sample) or "Somatic SV annotation" stage (if the sample was uploaded as a tumor sample) is launched. If any of the tasks listed below fail, the sample analysis is stopped.
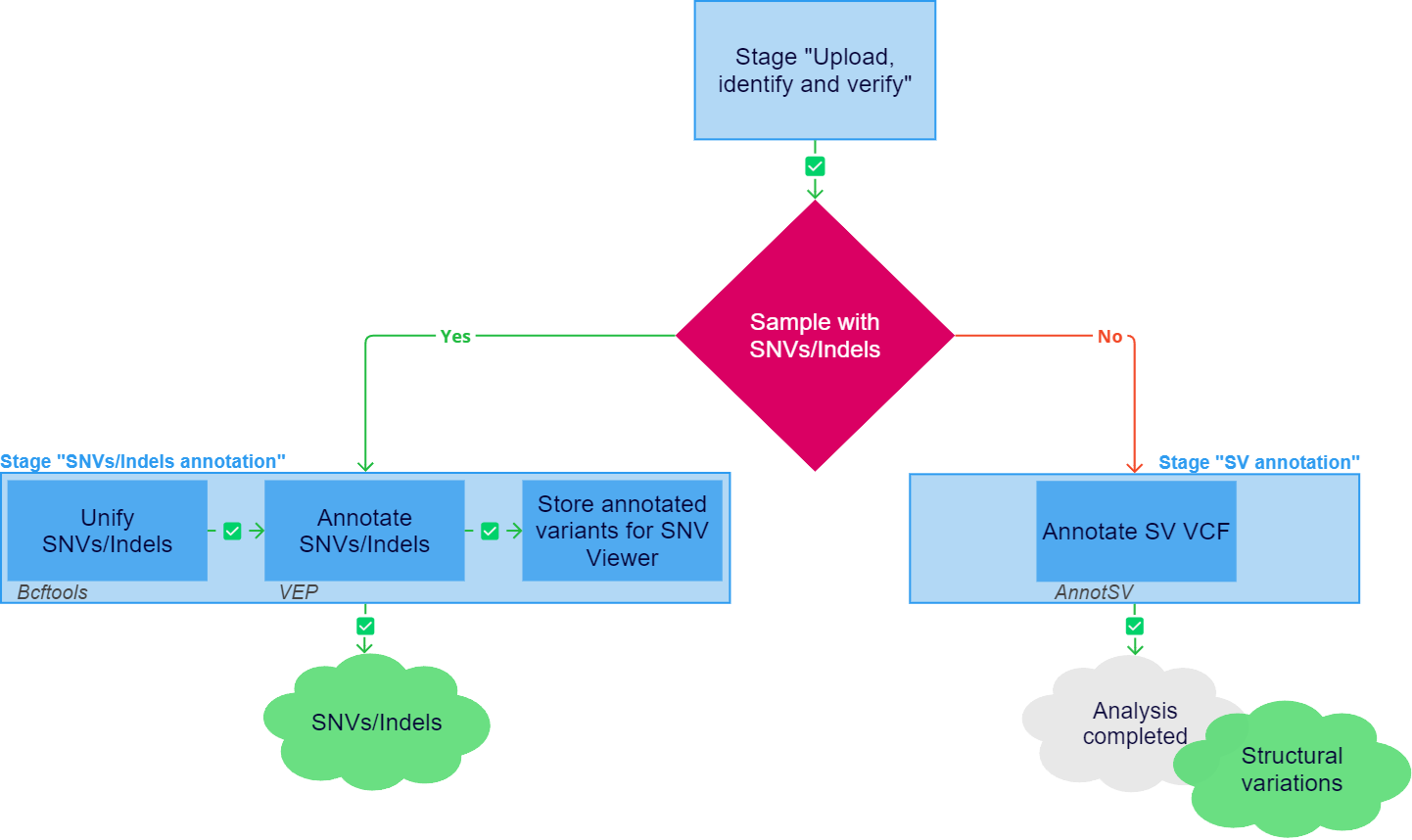
Tasks of the "SNVs/Indels annotation" stage:
- Unify SNVs/Indels:
- Sorting a VCF file using GNU sort by chromosome names.
- bcftools reheader modifies header of VCF file and changes sample names.
- GATK FixVCFHeader replaces or fixes a VCF header.
- bcftools norm checks if reference alleles in the file match the reference; splits multiallelic sites into biallelic records; outputs only the first instance if a record is present multiple times.
- Compressing a file into a GZIP archive using bgzip. The resulting file can be downloaded in the "Result files" section in the "Unify SNVs/Indels" task details ("Download VCF_GZ"). You can also open this file in the IGV browser by clicking on the "Open in IGV Browser" link.
- Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download VCF_TBI").
If the sample analysis workflow includes oligogenic/polygenic risk scores calculation, pharmacogenetic calculation and/or ancestry analysis, then the "Genomic predictions" stage will be launched in parallel with annotation after the "Unify SNVs/Indels" task.
- Annotate SNVs/Indels:
Annotation of SNVs/Indels in the file with data from the RefSeq, 1000 Genomes, dbNSFP, dbSNP, gnomAD 3, gnomAD 4, ClinVar, CADD, SpliceAI, ENIGMA databases.
Determination of the effect of SNVs/Indels on genes, transcripts, and protein sequence, as well as regulatory regions, using Ensembl Variant Effect Predictor (VEP):
PolyPhen predicts possible impact of an amino acid substitution on the structure and function of a protein using straightforward physical and comparative considerations.
A flag indicating if the transcript is the canonical transcript for the gene is added.
Allele number from VCF input is identified.
Affected exon and intron numbering is added.
HGVS nomenclature based on Ensembl stable identifiers is added.
Upstream variant consequences are determined if the upstream (5') distance between a variant and a transcript is more than 2000 bp. Downstream variant consequences are determined if the downstream (3') distance between a variant and a transcript is more than 1000 bp.
The resulting file with raw annotated SNVs/Indels in TSV format can be downloaded in the "Result files" section in the "Annotate SNVs/Indels" task details ("Download Raw annotated TSV"). You can also open it in Google Sheets.
The resulting file with annotated SNVs/Indels without duplicates in TSV format can be downloaded in the same section ("Download All variants TSV"). You can also open it in Google Sheets. The same file in CSV format can be downloaded from the same place ("Download All variants CSV").
Converting TSV results to VCF.
GATK FixVCFHeader replaces or fixes a VCF header.
The file is compressed into a GZIP archive using bgzip. The resulting file can be downloaded in the "Result files" section in the "Annotate SNVs/Indels" task details ("Download All variants VCF_GZ"). You can also open this file in the IGV browser by clicking on the "Open in IGV Browser" link.
The file is indexed using tabix. The resulting index file can be downloaded in the same section ("Download All variants VCF_TBI").
Calculating variants' statistic.
- Store annotated variants for SNV Viewer: saving results for display in SNV Viewer, i.e. an embedded service for viewing and analyzing variants, and adding information about the variant occurrence in other samples of the user.
After the "Annotate SNVs/Indels" stage, the analysis can proceed with report generation.
The "SV annotation" stage includes one task "Annotate SV VCF":
- Annotation and ranking of structural variations using AnnotSV.
The resulting file with structural variations in TSV format can be downloaded in the "Result files" section in the "Annotate SV VCF" task details ("Download Filtered variants TSV"). You can also open it in Google Sheets. The same file in CSV format can be downloaded from the same place ("Download Filtered variants CSV"). - Compressing a file into a GZIP archive using bgzip. The resulting file can be downloaded in the same section ("Download Filtered variants VCF_GZ"). You can also open this file in the IGV browser by clicking on the "Open in IGV Browser" link.
- Indexing a file using tabix. The resulting index file can be downloaded in the same section ("Download Filtered variants VCF_TBI").
The "SV annotation" stage completes the sample analysis.
info
If you want to add a track with SNVs/Indels or structural variations discovered by the analysis of the sample you uploaded to Genomenal to your desktop IGV, you can do so via a link. Open the details of the necessary task ("Unify SNVs/Indels", "Annotate SNVs/Indels", "Annotate SV VCF") and do the following:
- Right-click the variant file link (depending on the selected task, this may be the link "Download VCF_GZ", "Download All variants VCF_GZ" or "Download Filtered variants VCF_GZ") and select the "Copy link address" option.
- Upload the track via URL to your desktop IGV, as described here.
- Right-click on the download link for the index file corresponding to the annotation file ("Download VCF_TBI", "Download All variants VCF_TBI" or "Download Filtered variants VCF_TBI") and select the "Copy link address" option.
- Add the index via URL to the corresponding field in IGV.
- Click "OK". Done! The track with SNVs/Indels or structural variations discovered in the sample is added to IGV.