SNV Viewer
Overview#
SNV Viewer is an embedded service for viewing and analyzing single-nucleotide variants (SNVs) and short insertions/deletions (Indels) discovered in the sample. Located on Main tab of the sample.
On the tumor sample page, you can find two SNV Viewers: one with somatic SNVs/Indels discovered in this sample and one with germline SNVs/Indels discovered in the control sample in the case of the tumor sample from a pair of tumor/normal samples or in the tumor sample itself, if it was analyzed without a control sample. In SNV Viewer with somatic mutations, you can also see SNVs/Indels from SNV Viewer with germline mutations if you change the value of the filter by origin from Somatic to Germline or clear filtering by it.

The non-tumor sample page presents SNV Viewer with germline SNVs/Indels only discovered in this sample:
Main Page#
After clicking on  , you
will see the main page of the service:
, you
will see the main page of the service:
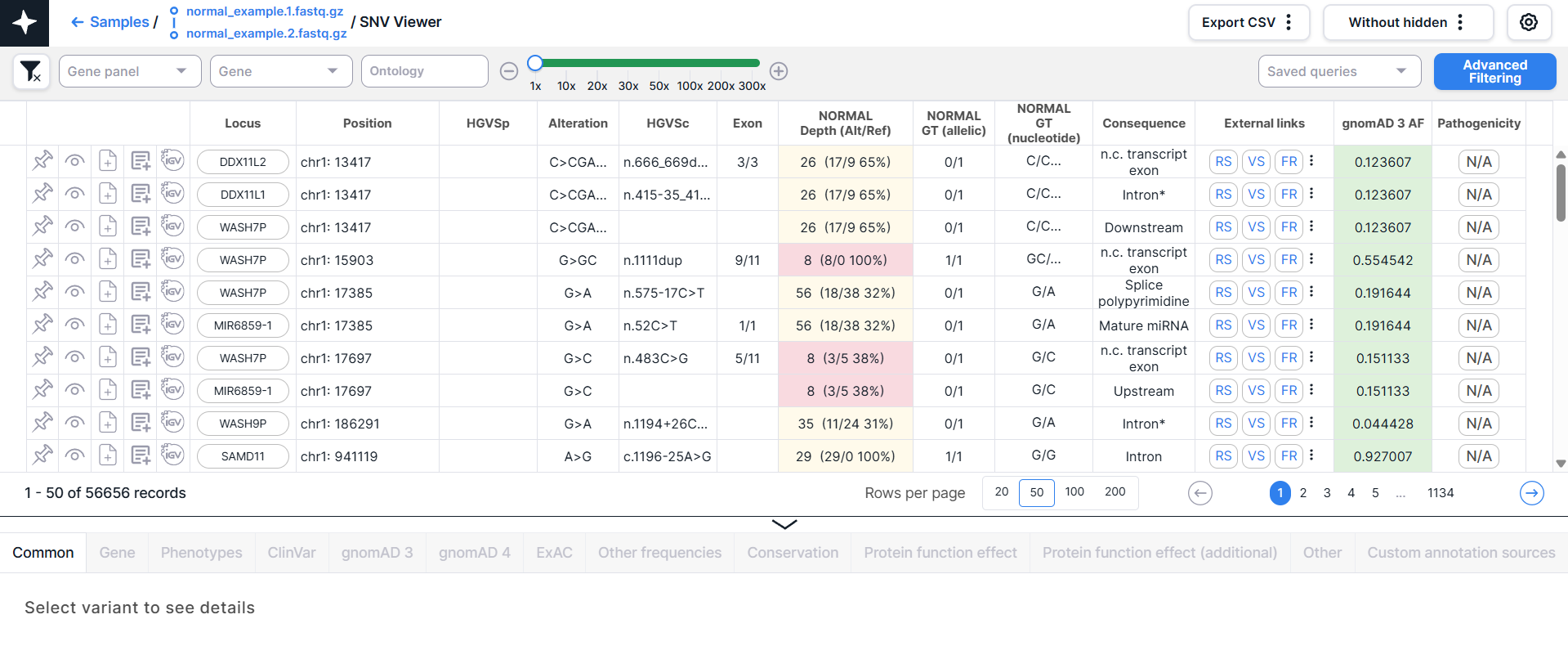
The main object of SNV Viewer is a table with variants - each variant is on a new line:

Below the table, above the variant details panel, there are:
- Information about the amount of variants in SNV Viewer: the order of variants displayed on the page (e.g., if the maximum amount of variants shown on the page is 50, then the order will be: 1-50, 51-100, etc.) and the total amount of variants (pinned variants are not taken into account). The amount of variants changes when filtering variants and displays the amount of variants that match the specified conditions.
- A component to choose the amount of variants displayed on the page
(excluding pinned variants):
 .
By default, 50 variants are displayed per page. To change the amount of variants displayed on the page to
20, 100 or 200, click on the corresponding option. Your choice will be remembered and will be valid for SNV Viewer
and CNV Viewer of all samples. Please note that the more variants shown on a page, the longer it will take to
load the page.
.
By default, 50 variants are displayed per page. To change the amount of variants displayed on the page to
20, 100 or 200, click on the corresponding option. Your choice will be remembered and will be valid for SNV Viewer
and CNV Viewer of all samples. Please note that the more variants shown on a page, the longer it will take to
load the page. - Pagination component:
 , which allows you to navigate between pages with variants. The number of the current page is highlighted in a blue circle. If you click on this number, the page will reload. To move to a page with a specific number, either click on that number, or click on
, which allows you to navigate between pages with variants. The number of the current page is highlighted in a blue circle. If you click on this number, the page will reload. To move to a page with a specific number, either click on that number, or click on  , enter the number and press the enter key or click outside the value field. To move to the page with previous number, click on
, enter the number and press the enter key or click outside the value field. To move to the page with previous number, click on  , and to open the next page, click on
, and to open the next page, click on  .
.
Variant Info#
Besides of the information presented in the table (see Table Columns' Description), for each variant there are several embedded modules with additional information:
- Variant detailed information panel. Click on the variant row to see a panel:
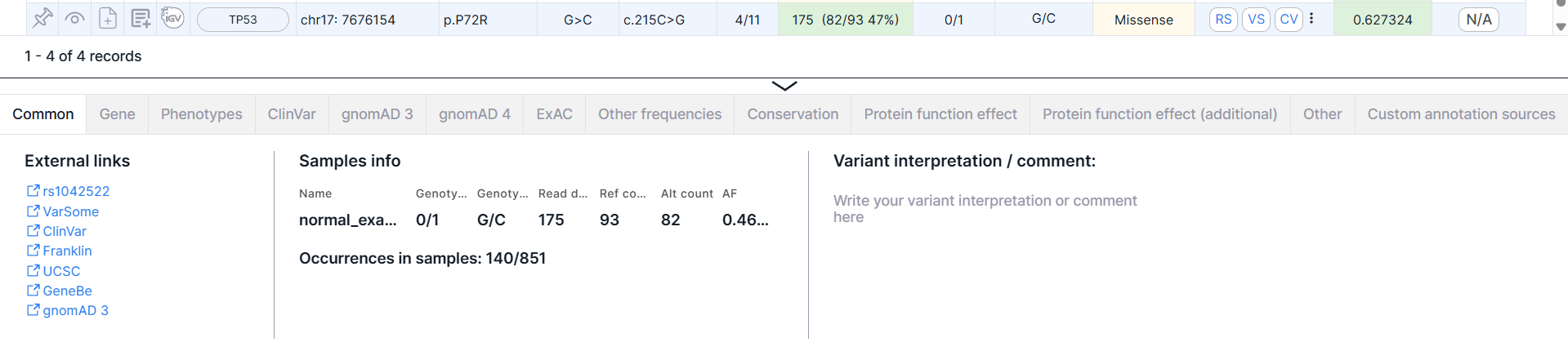
You can read more about the panel and its tabs in the corresponding section.
- Variant details page. To open the page,
click on
 in the variant row.
On the page, you can add a variant interpretation,
determine a variant pathogenicity, and
find more information about the variant. The page may include the following sections:
in the variant row.
On the page, you can add a variant interpretation,
determine a variant pathogenicity, and
find more information about the variant. The page may include the following sections:
- Detailed variant annotation.
- Sample and Patient info.
- Information about occurrences of the variant in other samples.
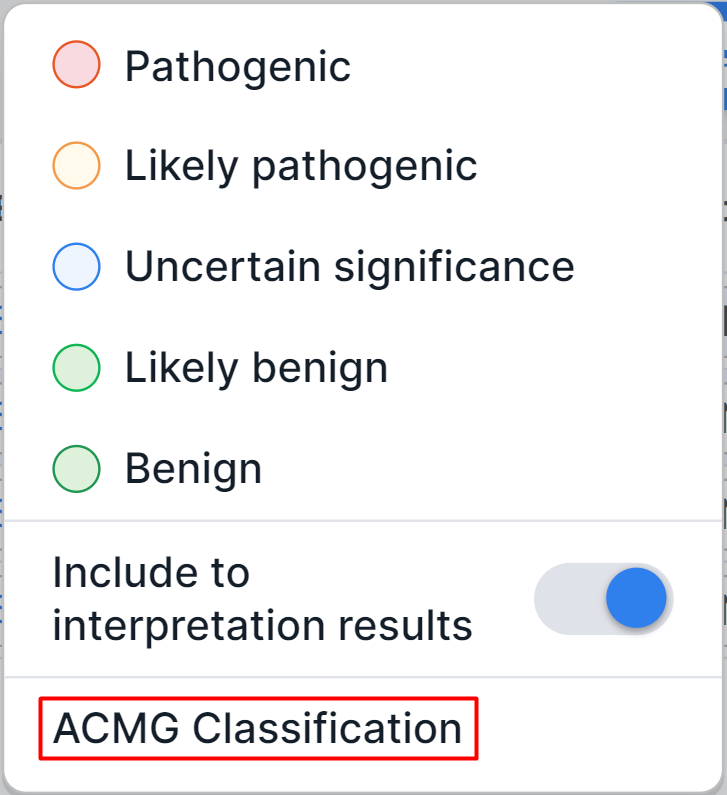
- ACMG classification: includes criteria for variant
interpretation developed by ACMG. You can read more about this page
in the corresponding section. To open
the page directly, click on the pathogenicity button
(
 if the variant pathogenicity has not
been previously determined) in the variant row ("Pathogenicity" column) and click on "ACMG Classification"
option in the dialog:
if the variant pathogenicity has not
been previously determined) in the variant row ("Pathogenicity" column) and click on "ACMG Classification"
option in the dialog:
- Scientific publications associated with the variant.
- Clinical guidelines.
The page is available if the variant is onco relevant.
To open page directly, click on
 in
the variant row ("Onco Relevance" column - can be added
in column edit mode).
in
the variant row ("Onco Relevance" column - can be added
in column edit mode). - Variant report.
- Integrative Genomics Viewer (IGV) is a module for visualization of variant on the genome.
Click on
 in the variant row to open the module.
in the variant row to open the module.
Pin Variants#
The variants you want can be pinned. Pinned variants are not affected by filtering and sorting the table and always remain at the top of the table. The visibility of pinned variants in the table can only be affected by filtering by origin (for example, pinned germline variants will not be presented in a table filtered by somatic origin). Pinned variants are sorted by the variant start position ("Position" column).
Click on ![]() in the variant line to pin it. The pinned variant will appear at the top of the table (the row highlighted in green):
in the variant line to pin it. The pinned variant will appear at the top of the table (the row highlighted in green):

To unpin a variant, click on the same button ![]() in the
variant row.
in the
variant row.
You can also pin or unpin a variant using keyboard shortcuts. To do this, select the variant row (the selected row is highlighted in blue) and press P on the keyboard.
note
Variant remains pinned when you exit the SNV Viewer page.
Hide Variants#
Variants can be hidden from SNV Viewer table. To do this,
click on ![]() in the variant row.
Hidden variants will only be available when a specific filter condition is applied.
in the variant row.
Hidden variants will only be available when a specific filter condition is applied.
Variant Visibility Filter: in the upper right corner of SNV Viewer page (regardless of variant filtering mode),
there is a variant visibility filter button:  (default state):
(default state):

- By default, only visible (not hidden) variants are shown in SNV Viewer table (filter option "Without hidden").
- To see in the table all variants discovered and annotated in the sample, click on the filter button
and select "Show all" option. Then the table will display all variants, including hidden ones.
The visible variants in the row have a button to hide:
 , while the hidden variants have a button to show:
, while the hidden variants have a button to show:  .
. - To see only hidden variants, click on the filter button and select "Hidden only" option.
note
Pinned variants are always visible. At the same time, they are displayed in the table under any condition of variant visibility filter.
To make a hidden variant visible, click on ![]() in its row.
in its row.
You can also hide or show a variant using keyboard shortcuts. To do this, select the variant row (the selected row is highlighted in blue) and press H on the keyboard.
note
When you pin a hidden variant, it not only becomes pinned in SNV Viewer table, but also becomes visible.
Include Variants to Report#
Variants included to report will be included in report blocks
of "SNVs/Indels selected by user for reports" type.
To include a variant to the report,
click on ![]() in the variant row. To remove a variant from the report, click the same button
in the variant row. To remove a variant from the report, click the same button ![]() in the variant row.
Variants included to report can also be removed from the report on the page of the report itself.
in the variant row.
Variants included to report can also be removed from the report on the page of the report itself.
You can also include a variant to report or remove a variant from report using keyboard shortcuts. To do this, select the variant row (the selected row is highlighted in blue) and press R on the keyboard.
info
When a variant is determined as pathogenic or likely pathogenic, it is automatically included to report.
Variant Significance Label#
You can interpret a variant by its significance (clinical or for your study, etc.) To do this,
add "Label" column in column edit mode (not
added by default). The column contains a label
button ![]() ("Undefined significance"
if the label value has not been defined).
("Undefined significance"
if the label value has not been defined).
To interpret a variant by significance, click on the label
button ![]() and select the required value
in the drop-down list:
and select the required value
in the drop-down list:

You can later redefine the significance in the same way or clear it by clicking on the label button and selecting "Clear" option:
![]()
As a result of the variant interpretation by significance, the label button will change color:
- High significance:

- Moderate significance:

- Low significance:

- Sequencing error:

Variants can be sorted by significance: from high significance to sequencing error by clicking on "Label" column name once, and vice versa by clicking on the name twice. In addition, variants can be filtered by significance:
- "Label" filter in advanced filtering mode;
- an additional filter "Label" in basic filtering mode (can be added on "Profile" page).
Filter and Sort Variants#
Filtering of variants in SNV Viewer is described in Basic Filtering and Advanced Filtering sections.
By default, the table is sorted by the variant start position ("Position" column). You can sort the table by the values of the following columns:
- default columns: "Locus", "Origin", "Position", "Exon", "Depth (Alt/Ref)", "Consequence", "gnomAD 3 AF", "Pathogenicity";
- all columns not added by default.
To sort a table by column values, click on the column name (click once to sort in ascending order,
or twice to sort in descending order). After that, you will see an arrow next to the column
name: ![]() (when sorting in ascending order) or
(when sorting in ascending order) or ![]() (when sorting in descending order).
(when sorting in descending order).
You can sort a table by 1-3 columns at the same time to group data by the same value in one column, and then sort another column within that group of equal values. The sort order will depend on the order in which you click on the corresponding column names. The sort arrow will be presented for the column that was sorted last.
note
Table sorting and filtering are saved when you exit the SNV Viewer page.
To reset sorting, use the clear all filters
button: ![]() .
Please note that this will also reset the filtering of variants.
.
Please note that this will also reset the filtering of variants.
The basic sorting orders are alphabetical for columns with text values (e.g. "Locus" column), or smallest to largest and vice versa for columns with numeric values (e.g. "gnomAD 3 AF" column). However, the sorting order is special for some columns:
- "Consequence" column: sorting from most significant consequence to least significant: Transcript ablation, Splice acceptor, Splice donor, Stop gained, Frameshift, Stop lost, Start lost, Transcript amplification, Inframe insertion, Inframe deletion, Missense, Protein altering, Splice region, Incomplete terminal codon, Start retained, Stop retained, Synonymous, Coding sequence, Mature miRNA, UTR5, UTR3, Non coding transcript exon, Intron, NMD transcript, Non coding transcript, Upstream, Downstream, TFBS ablation, TFBS amplification, TF binding site, Regulatory region ablation, Regulatory region amplification, Feature elongation, Regulatory region variant, Feature truncation, Intergenic variant, Splice donor 5th base variant, Splice donor region variant, Splice polypyrimidine tract variant, Unknown. Composite values (containing multiple consequences) are sorted by the least significant value in the list.
- "Pathogenicity" column: sorting from pathogenic to benign variant: Pathogenic, Likely pathogenic, Conflicting interpretations of pathogenicity, Uncertain significance, Likely benign, Benign.
- "Impact" column (not added by default, values are described here): sorting from high to low impact: HIGH, MODERATE, LOW, MODIFIER.
- "Genome position" column (not added by default): sorting by the significance of the variant location in the genome: Exonic, Intronic, Intergenic.
- "Label" column (not added by default, described here):
sorting from significant to insignificant
variants: (high significance), (moderate significance), (low significance), (sequencing
error).
- "Caller Filters" column (not added by default, described here): sorting from variants which didn't pass filtration to variants satisfying all filters: FAIL, N/A, PASS.
- "ClinVar" column (not added by default, described here): sorting from unconditionally significant variants to insignificant ones:
- Pathogenic;
- Pathogenic, low penetrance;
- Likely pathogenic;
- Likely pathogenic, low penetrance;
- Composite values including "Pathogenic" or "Pathogenic, low penetrance";
- Composite values including "Likely pathogenic" or "Likely pathogenic, low penetrance";
- Uncertain significance;
- Conflicting interpretations of patogenicity / Conflicting data from submitters;
- Established risk allele;
- Likely risk allele;
- Uncertain risk allele;
- Risk factor;
- Affects;
- Drug response;
- Association;
- Confers sensitivity;
- Protective;
- Likely benign;
- Association not found;
- Benign;
- Other;
- Not provided.
Composite values (other than those that include the values "Pathogenic", "Pathogenic, low penetrance", "Likely pathogenic", or "Likely pathogenic, low penetrance") are sorted by the most significant value from the list.
- "ENIGMA" column (not added by default, described here): sorting from significant to insignificant variants: Pathogenic, Likely pathogenic, Likely benign, Benign, Not provided. Composite values are sorted by the most significant value from the list.
Keyboard Shortcuts in SNV Viewer#
In SNV Viewer, you can interact with variants using keyboard shortcuts. To use keyboard shortcuts, click on a variant row, or press one of keys: S, W, ⬇️, or ⬆️ to select the first variant in the table.
Available keyboard shortcuts
| Press this key | To do this |
| S or ⬇️ | Move to the row of the next variant (or the first variant in the table if no variant was selected) |
| W or ⬆️ | Move to the row of the previous variant (or the first variant in the table if no variant was selected) |
| P | Pin/unpin variant |
| H | Hide/show variant |
| R | Include variant to report or remove it from report |
Export Variants#
SNV Viewer allows to export variants in CSV format:
- Export of all variants: click on
 and select
"All" option:
and select
"All" option:

- Export of only filtered variants: filter the needed variants by
Basic or Advanced
filtering, then click on and select
"All" option:
- Export of only pinned variants: pin
the needed variants, then click on and select
"Pinned only" option:

note
If no variant has been pinned in SNV Viewer, then "Pinned only" option will not be in the drop-down list of export options.
- Export of only modified variants: click
on and select "Modified only" option:
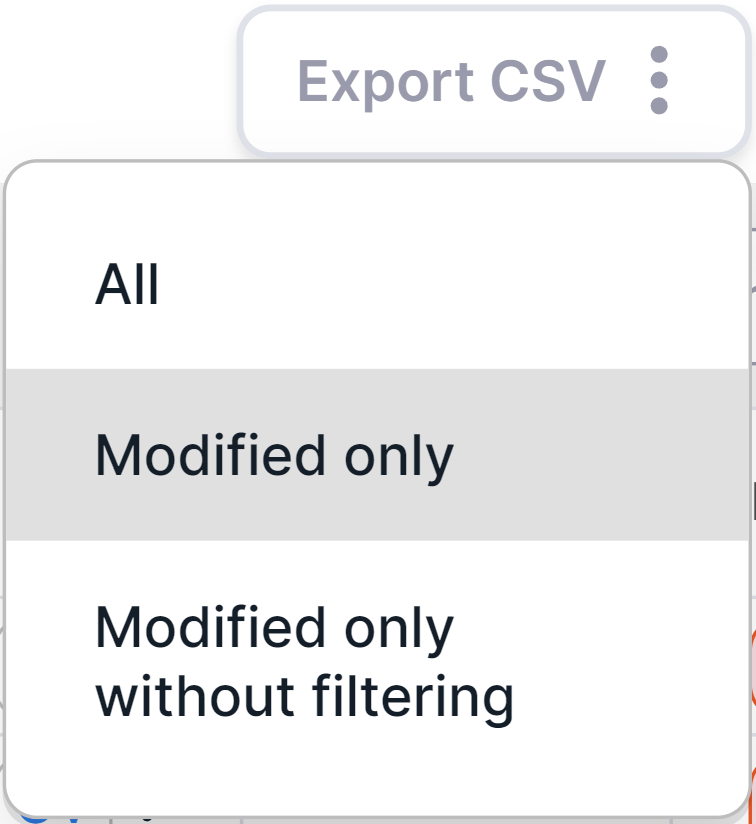
Modified variants are variants for which any of the following has been done:
- a variant is pinned in SNV Viewer table;
- a variant is included to report;
- interpretation was added for a variant;
- pathogenicity was determined for a variant;
- a variant is interpreted by significance.
note
If SNV Viewer table is filtered by some condition, then the table of exported modified variants will include only those modified variants that match this condition. An exception is pinned variants, whose visibility in the table can only be affected by filtering by origin.
To export all modified variants without filtering,
click on and select
"Modified only without filtering" option:
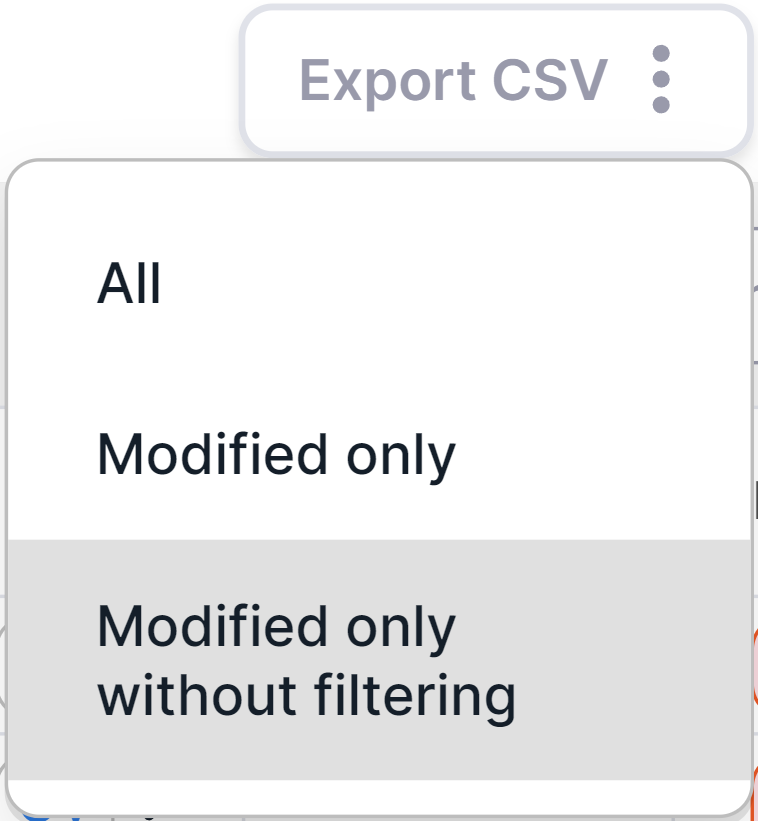
Table Columns' Description#
By default, the table has the following columns (the set and order of the columns can be changed - how to do it is described here):
- Locus is the common name of the gene in which the variant is located. If the gene is unknown,
the locus is designated as
 . You can copy the gene name to the clipboard by moving the cursor over the column value and clicking on
. You can copy the gene name to the clipboard by moving the cursor over the column value and clicking on  .
. - Origin is a mutation type (somatic or germline).
info
"Origin" column is hidden in SNV Viewer with germline mutations and in SNV Viewer with somatic mutations if "Origin is Somatic" filter is on.
- Position is a coordinate of the variant in the genome (chromosome + start position).
You can copy the variant position to the clipboard by moving the cursor over the column value and
clicking on .
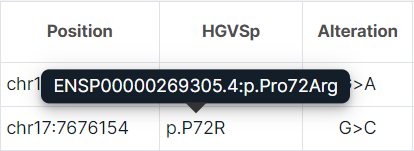
- HGVSp is an amino acid substitution using the HGVS notation (“p.” prefix (protein) + reference amino acid + amino acid position in protein + new amino acid resulting from the substitution). Amino acids are written in one letter code. When hovering over the value of the "HGVSp" column for a variant, you can see the transcript which the substitution was predicted for and HGVSp with amino acids in the three letter code:
note
If you prefer to use amino acid three letter coding, add the "HGVSp (long)" column in the Edit Table Columns window and hide the "HGVSp (short)" column.
- Alteration is a nucleotide substitution (reference allele > alternative allele).
- HGVSc is a nucleotide substitution using the HGVS notation (genomic position of the substituted nucleotide + reference allele > alternative allele). Starts with a prefix: “c.” (coding; for a substitution in the coding sequence) or “n.” (non-coding; for a substitution in the non-coding sequence). When hovering over the value of the "HGVSc" column for a variant, you can see the transcript which the substitution was predicted for (the transcript IDs from Ensembl (ENSTxxxxxxxxxxx) and RefSeq (NM_) are given):
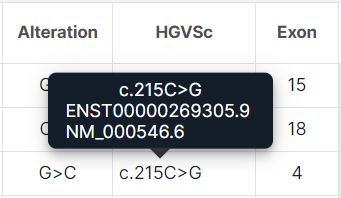
- Exon is the exon number in the transcript which is affected by the variant.
- Depth (Alt/Ref) is a sequencing depth; the total number of reads of the sequence overlapping the variant position for this sample. Alt is the alternative allele reads count, Ref is the reference allele reads count. Also the percentage of the share of the alternative allele reads count among all reads (Alt/Depth) is shown.
The column cells are colored depending on the depth value:
- green if depth > 60;
- yellow if 20 ≤ depth ≤ 60;
- red if depth < 20.
- GT is a genotype; allelic values (0 - reference allele, 1 - first alternative allele, 2 - second alternative allele etc.), divided by ”/” (for unphased genotype) or “|” (for phased genotype).
- Consequence is the effect of the variant on genes.
Consequences of variants in the annotation#
| Consequence | Description | Impact* |
| Coding sequence | The variant hits a coding sequence | MODIFIER |
| Downstream | Downstream of a gene | MODIFIER |
| Feature elongation | A sequence variant that causes the extension of a genomic feature | MODIFIER |
| Feature truncation | A sequence variant that causes the reduction of a genomic feature | MODIFIER |
| Frameshift | Insertion or deletion causes a frame shift | HIGH |
| Incomplete terminal codon | A sequence variant where at least one base of the final codon of an incompletely annotated transcript is changed | LOW |
| Inframe deletion | One or many codons are deleted | MODERATE |
| Inframe insertion | One or many codons are inserted | MODERATE |
| Intergenic variant | The variant is in an intergenic region | MODIFIER |
| Intron | The variant is located in the intron | MODIFIER |
| Mature miRNA | A transcript variant located with the sequence of the mature miRNA | MODIFIER |
| Missense | Variant causes a codon that produces a different amino acid | MODERATE |
| NMD transcript | The variant is located in the transcript susceptible to nonsense-mediated decay | MODIFIER |
| Non coding transcript | The variant is located in the non-coding transcript | MODIFIER |
| Non coding transcript exon | A sequence variant that changes non-coding exon sequence in a non-coding transcript | MODIFIER |
| Protein altering | A variant which is predicted to change the protein encoded in the coding sequence | MODIFIER |
| Regulatory region ablation | Deletion of a regulatory region | HIGH |
| Regulatory region amplification | Amplification of regulatory region | HIGH |
| Regulatory region variant | The variant hits a regulatory feature | MODIFIER |
| Splice acceptor | The variant hits a splice acceptor site | HIGH |
| Splice donor | The variant hits a splice donor site | HIGH |
| Splice donor 5th base variant | A variant causes a change at the 5th base pair after the start of the intron in the orientation of the transcript | LOW |
| Splice donor region variant | A variant falls in the region between the 3rd and 6th base after splice junction (5' end of intron) | LOW |
| Splice polypyrimidine tract variant | A variant falls in the polypyrimidine tract at 3' end of intron between 17 and 3 bases from the end (acceptor -3 to acceptor -17) | LOW |
| Splice region | A sequence variant in which a change has occurred within the region of the splice site, either within 1-3 bases of the exon or 3-8 bases of the intron | LOW |
| Start lost | Variant causes start codon to be mutated into a non-start codon | HIGH |
| Start retained | Variant causes start codon to be mutated into another start codon | LOW |
| Stop gained | Variant causes a STOP codon | HIGH |
| Stop lost | Variant causes stop codon to be mutated into a non-stop codon | HIGH |
| Stop retained | Variant causes stop codon to be mutated into another stop codon | LOW |
| Synonymous | Variant causes a codon that produces the same amino acid | LOW |
| TF binding site | Transcription factor binding site (TFBS) | MODIFIER |
| TFBS ablation | Deletion of a TFBS | HIGH |
| TFBS amplification | Amplification of a TFBS | HIGH |
| Transcript ablation | Deletion of a transcript | HIGH |
| Transcript amplification | A feature amplification of a region containing a transcript | HIGH |
| Upstream | Upstream of a gene | MODIFIER |
| UTR3 | A variant hits 3'UTR region | MODIFIER |
| UTR5 | A variant hits 5'UTR region | MODIFIER |
Cells of "Consequence" column are colored depending on the impact*:
- red if the impact is HIGH;
- yellow if the impact is MODERATE;
- no color if the impact is LOW or MODIFIER.
Values that include multiple consequences are indicated with an asterisk. You can see the full list of consequences by hovering over a cell:
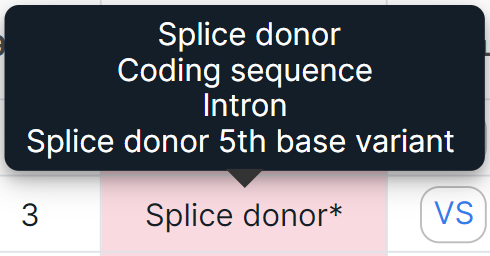
*Impact is the predicted effect of the variant on the protein. The column is not added to the table by default, it can be added in Edit Table Columns window.
Impacts of variants in the annotation#
| Impact | Description |
| HIGH | The variant has high (disruptive) impact in the protein, probably causing protein truncation, loss of function or triggering nonsense mediated decay |
| MODERATE | The variant might change protein effectiveness |
| LOW | The variant is mostly harmless or unlikely to change protein behavior |
| MODIFIER | Usually variants affecting non-coding genes, where predictions are difficult or there is no evidence of impact |
The "Impact" column cells are colored:
- red if the impact is HIGH;
- yellow if the impact is MODERATE;
- no color if impact is LOW or MODIFIER.
- External links are links to pages with variant information
in dbSNP:
 , VarSome:
, VarSome:  , ClinVar:
, ClinVar:  , COSMIC:
, COSMIC:  (if COSMIC database was uploaded as a custom annotation), Franklin:
(if COSMIC database was uploaded as a custom annotation), Franklin:  , UCSC:
, UCSC:  , gnomAD 3:
, gnomAD 3:  , RUSeq:
, RUSeq:  (if RUSeq database was uploaded as a custom annotation). There may be up to three buttons maximum in a cell. If the variant is found in more than three databases, then there will be three dots
(if RUSeq database was uploaded as a custom annotation). There may be up to three buttons maximum in a cell. If the variant is found in more than three databases, then there will be three dots  in the cell.
By clicking on these dots, you can see all the other links:
in the cell.
By clicking on these dots, you can see all the other links:

- gnomAD 3 AF is the allele frequency according to the gnomAD v3 database.
The column cells are colored depending on the allele frequency value:
- green if AF ≥ 0.015;
- yellow if 0.01 ≤ AF < 0.015;
- red if AF < 0.01.
- Pathogenicity is the variant pathogenicity, which can be determined either manually, by ACMG criteria, or
according to the pathogenicity base. If you have not uploaded the pathogenicity database
or have not determined the variant pathogenicity, then the column
value will be (No interpretation).
Description of Additional Table Columns#
In addition to the columns described above, other annotation columns can be added to SNV Viewer table as described here. The columns with special features are described below:
- "Label" column, in which the variant significance can be determined, is described above.
- "PG" column contains information about the phasing group of the variant.
It is called "Phasing group status" ("Common" section)
in Edit Table Columns window.
The phasing group (i.e. a merging of homozygous variants located within a specific genomic interval)
is indicated by
 . The variants that are part of phasing group with discordant amino acid substitutions indicated by
. The variants that are part of phasing group with discordant amino acid substitutions indicated by  and the variants that are part of phasing group with concordant amino acid substitution indicated by
and the variants that are part of phasing group with concordant amino acid substitution indicated by  .
. - "Onco Relevance" column contains a
button if the variant
is onco-relevant, i.e. associated with the development of cancer.
The button opens "Clinical Guidelines" tab of the
variant details page.
- "Caller Filters" column contains information about the variant quality:
1. PASS - site contains at least one allele that satisfies all filters. The cell is colored green.
2. N/A - the column value is unknown (dot in VCF file). The cell is colored yellow.
3. FAIL - the variant does not satisfy one or more GATK filters. By hovering over a cell, you can see these filters. The cell is colored red. - "ClinVar" column contains information about the clinical significance of the variant
in ClinVar database.
It is called "Clinical significance" ("ClinVar" section)
in Edit Table Columns window.
Column values are presented
as icons:
 - Affects;
- Affects; - Association not found;
- Association not found; - Association;
- Association; - Benign;
- Benign; - Conflicting interpretations of patogenicity or Conflicting data from submitters;
- Conflicting interpretations of patogenicity or Conflicting data from submitters; - Drug response;
- Drug response; - Likely benign;
- Likely benign; - Likely pathogenic or Likely pathogenic, low penetrance;
- Likely pathogenic or Likely pathogenic, low penetrance; - Likely risk allele;
- Likely risk allele; - Not provided;
- Not provided; - Other;
- Other; - Pathogenic or Pathogenic, low penetrance;
- Pathogenic or Pathogenic, low penetrance; - Protective;
- Protective; - Established risk allele;
- Established risk allele; - Risk factor;
- Risk factor; - Confers sensitivity;
- Confers sensitivity; - Uncertain risk allele;
- Uncertain risk allele; -
Uncertain significance.
-
Uncertain significance.
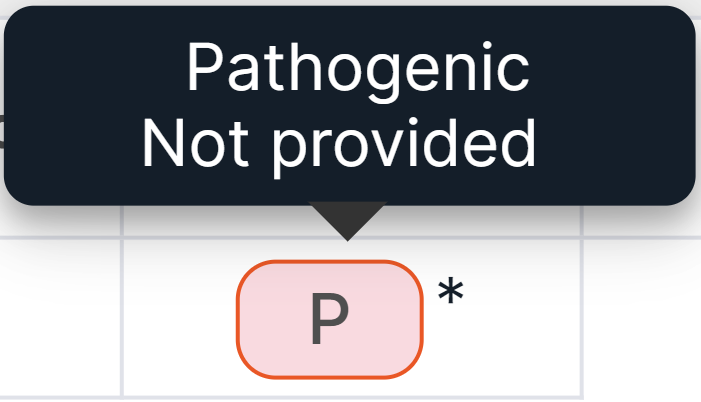
You can see the full value by hovering over the icon. Values that include multiple significances are indicated with an asterisk, and the icon in this case indicates the first value from the list:

- "ENIGMA" column contains information about clinical significance of the variant if it's located in the known or
suspected breast and/or ovarian predisposition genes (such as BRCA1, BRCA2) determined
by ENIGMA consortium.
It is called "ENIGMA clinical significance" ("Other" section)
in Edit Table Columns window.
Column values are presented
as icons: - Pathogenic; - Likely pathogenic; - Likely pathogenic; - Benign; -
Not provided.
You can see the full value by hovering over the icon. Values that include multiple significances are indicated with an asterisk, and the icon in this case indicates the first value from the list: