Variation Details Panel
Click on a variation row to see a panel with detailed information about it below the table. The panel is different for full and split rows. Panel with detailed information about the variation full length:
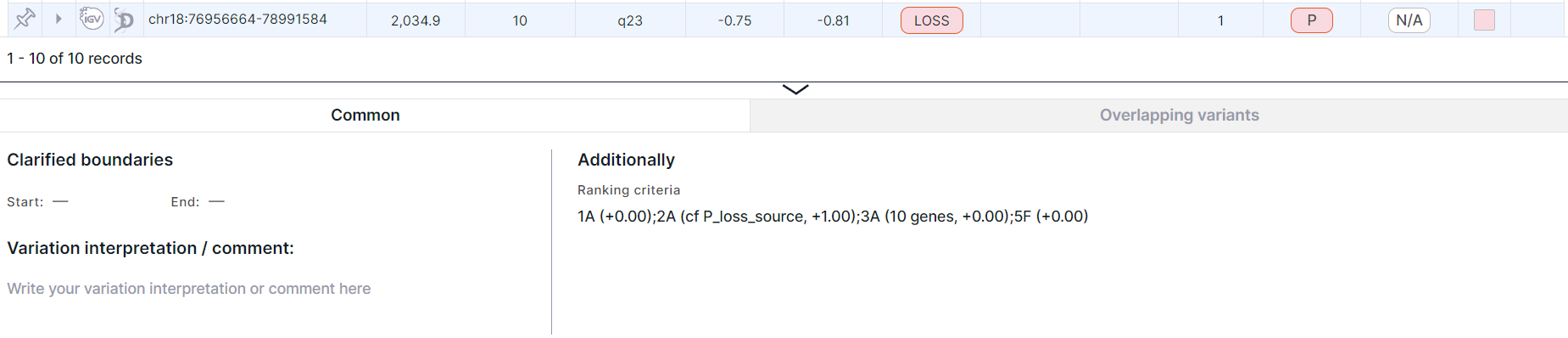
Panel with detailed information about each gene overlapped by the variation:
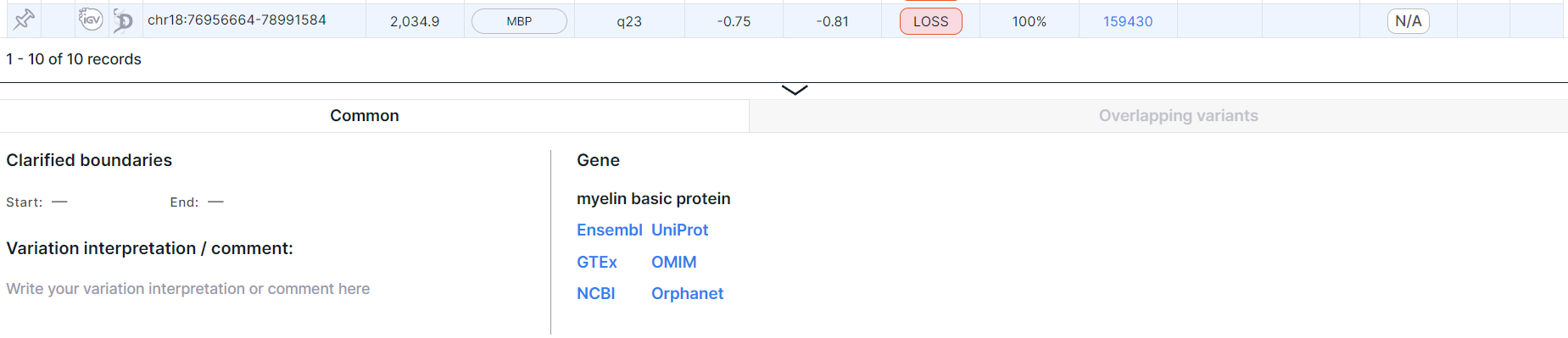
The panel can be resized by hovering over the top border of the panel and dragging down or up.
You can also collapse or expand the panel by
clicking on ![]() or
or ![]() , respectively.
, respectively.
info
On "Profile settings" page, you can adjust whether the variation details panel is expanded or collapsed in CNV Viewer by default.
The panel is divided into "Common" and "Overlapping variants" tabs.
On the panel, only those tabs are visually highlighted that include at least one non-empty variation annotation
field. For example, the "Overlapping variants" tab filled with data looks
like this:  , and empty - like this:
, and empty - like this: ![]() .
As you move between variations in the table, the selected panel tab is retained.
.
As you move between variations in the table, the selected panel tab is retained.
Common#
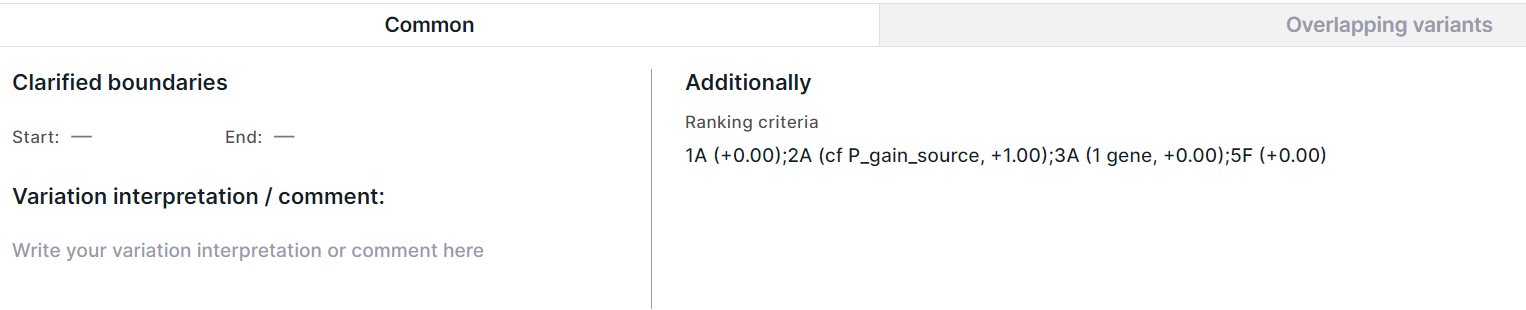
The "Common" tab looks different for full and split rows. The tab with common information about the variation full length looks like this:
The tab with common information about each gene overlapped by the variation looks like this:

The tab contains common information about the variation and may include the following sections:
- Clarified boundaries;
- Variation interpretation;
- Additionally (only for full rows);
- Gene (only for split rows);
- OMIM Gene-Phenotype Relationships (only for split rows if you are granted access to OMIM database).
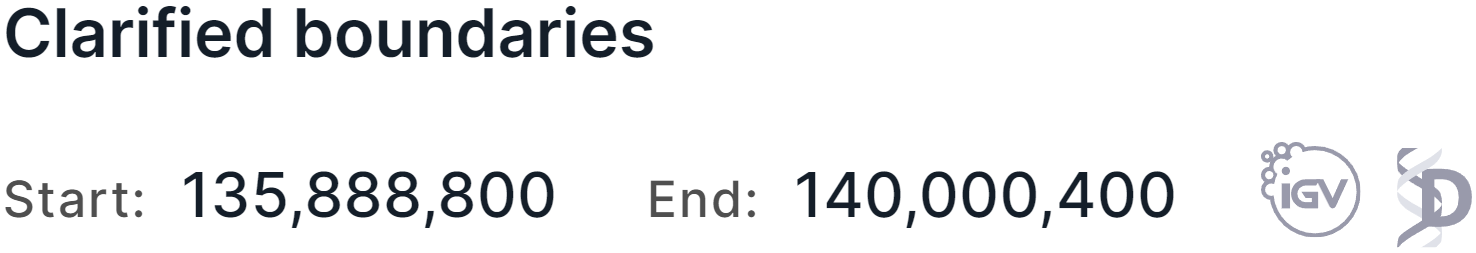
Clarified boundaries#
In the section, you can specify the start and end positions of the variation by entering them in the
corresponding “Start” and “End” fields. You can only enter numbers without any additional characters.
After clarifying the boundaries of variation, buttons to open this region in Integrative Genomics Viewer
(IGV) ![]() and Decipher genome browser
and Decipher genome browser ![]() will appear:
will appear:
Variation interpretation / comment#
A variation interpretation is a comment with important information about the variation added by the user. You can add or edit the interpretation by clicking on the input box, entering text and clicking outside the input box to save the interpretation:
Additionally#
For full rows, the ranking criteria are provided, i.e. the decision criteria that explain the ranking score provided by AnnotSV. Detailed guidelines for evaluating the ranking criteria are available in the file AnnotSV_Scoring_Criteria.xlsx (see Table 1 for duplications and Table 2 for deletions). Ranking score is the classification of variation pathogenicity provided by AnnotSV and based on ACMG and ClinGen recommendations1. According to the ranking score, the ACMG class is assigned:
- Variations with a ranking score ≥ 0.99 are assigned class 5 - pathogenic;
- Variations with a ranking score from 0.90 to 0.98 are assigned class 4 - likely pathogenic;
- Variations with a ranking score from 0.89 to −0.89 are assigned class 3 - uncertain significance;
- Variations with a ranking score from −0.90 to −0.98 are assigned class 2 - likely benign;
- Variations with a ranking score ≤ −0.99 are assigned class 1 - benign.
Gene#
For split rows representing each gene overlapped by the variation, the information about this gene is provided:
- Official full name. When hovering over the gene name, you can see from which databases it was taken:

- Links to pages with information about the gene in various databases (Ensembl, UniProt, GTEx, OMIM, NCBI, Orphanet).
OMIM Gene-Phenotype Relationships#
Access to data from OMIM database inside Genomenal is provided if the user has a license to access the database. If you have been granted access to OMIM, for split rows, there will be information on the phenotypes associated with the gene overlapped by the variation on the right side of the "Common" tab:
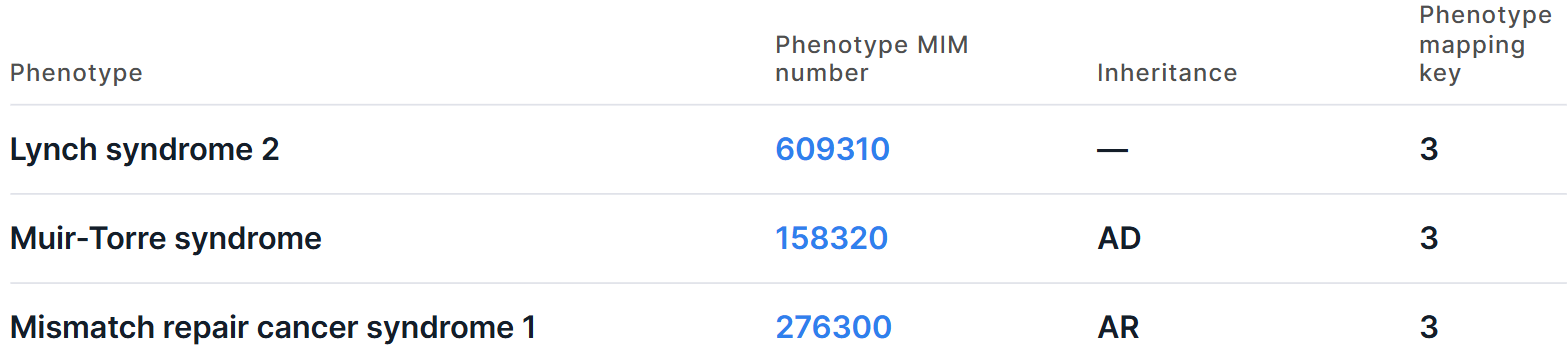
The information is presented in the form of a table with the following columns:
- Phenotype is the full name of the phenotype associated with the gene. For some phenotypes, there is a question mark before the name (e.g. "?Bleeding disorder, platelet-type, 22"), which indicates that the relationship between the phenotype and gene is provisional. Some phenotypes are enclosed in square brackets (e.g. "[Urate oxidase deficiency]"), which indicates mainly genetic variations that lead to apparently abnormal laboratory test values. Some phenotypes are enclosed in curly brackets (e.g. "{Prostate cancer/brain cancer susceptibility, somatic}"), which indicates mutations that contribute to susceptibility to multifactorial disorders or to susceptibility to infection.
- Phenotype MIM number is a phenotype ID in OMIM database. You can open the phenotype page by clicking on the link.
- Inheritance is a pattern of inheritance phenotype: AD - autosomal dominant; AR - autosomal recessive; DD - digenic dominant; DR - digenic recessive; IC - isolated cases; Mi - mitochondrial; Mu - multifactorial; PADom - pseudoautosomal dominant; PARec - pseudoautosomal recessive; SomMos - somatic mosaicism; SMu - somatic mutation; XL - X-linked; XLD - X-linked dominant; XLR - X-linked recessive; YL - Y-linked.
- Phenotype mapping key is an OMIM phenotype map key (the map displays relationships of the phenotype with associated phenotypes, phenotypic series and genes): 1 - the phenotype is placed on the map due to its association with a gene, but the underlying defect is not known; 2 - the phenotype has been placed on the map by statistical methods, no mutation has been found; 3 - the molecular basis of the disorder is known; 4 - a contiguous gene duplication or deletion syndrome in which multiple genes are involved.
Overlapping variants#
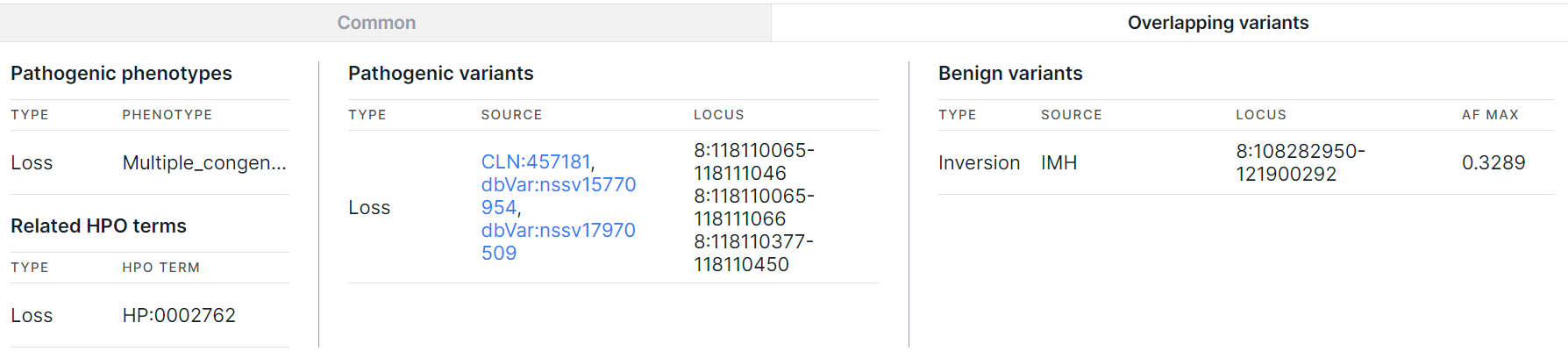
The tab contains information about phenotypes and HPO terms associated with the pathogenic loss, gain, or insertion genomic regions completely overlapped with the variation, as well as information about pathogenic and benign variants that overlap with the variation.
Pathogenic phenotypes#
The phenotypes are presented as a table with the following columns:
- Type is a copy number variation type: Loss (deletion), Gain (duplication), or Insertion.
- Phenotype is the name of the phenotype of the pathogenic loss, gain or insertion genomic regions completely overlapped with the variation. When you hover over a column value, you can see the full name of the phenotype.
Related HPO terms#
The HPO terms are presented as a table with the following columns:
- Type is a copy number variation type: Loss (deletion), Gain (duplication), or Insertion.
- HPO term is a term from Human Phenotype Ontology (HPO) describing the pathogenic loss, gain or insertion genomic regions completely overlapped with the variation.
Pathogenic variants#
Pathogenic overlapping variants are presented as a table with the following columns:
- Type is a copy number variation type: Loss (deletion), Gain (duplication), or Insertion.
- Source is an origin of the pathogenic loss, gain or insertion genomic regions completely overlapped with the variation. Possible sources: dbVar, ClinVar (CLN), ClinGen (TS3 for gain or HI3 for loss), disease-associated gene name from OMIM (morbid).
- Locus is the coordinates of the pathogenic loss, gain or insertion genomic regions completely overlapped with the variation (chromosome:start-end).
Benign variants#
Benign overlapping variants are presented as a table with the following columns:
- Type is a copy number variation type: Loss (deletion), Gain (duplication), Insertion, or Inversion.
- Source is an origin of the benign loss, gain, insertion or inversion genomic regions completely overlapped with the variation. Possible sources: gnomAD, ClinVar (CLN), ClinGen (TS40 for gain or HI40 for loss), DGV (dgv, nsv or esv), DDD, 1000 genomes (1000g), Ira M. Hall’s lab (IMH), Children’s Mercy Research Institute (CMRI).
- Locus is the coordinates of the benign loss, gain, insertion or inversion genomic regions completely overlapped with the variation (chromosome:start-end).
- AF MAX is the maximum allele frequency (AF) of the reported benign loss, gain, insertion or inversion genomic regions completely overlapped with the variation.
- Riggs E.R., Andersen E.F., Cherry A.M. et al. Technical standards for the interpretation and reporting of constitutional copy number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22, 245-257 (2020)↩