Геномные предсказания
После успешного выполнения стадии "Предварительная обработка для выявления вариантов" для образцов неопухолевой ткани может запуститься пайплайн геномных предсказаний, если в анализ была включена хотя бы одна из следующих задач: "Вычисление олигогенных рисков", "Вычисление полигенных рисков", "Расчет фармакогенетики" или "Анализ происхождения".
При ошибке выполнения любой из перечисленных ниже задач анализ образца останавливается. Однако, если в анализ включены контроль качества и импутация и если образец не удовлетворяет критериям контроля качества, то выполнение стадии "Геномные предсказания" останавливается, но анализ образца может продолжиться генерацией отчётов.
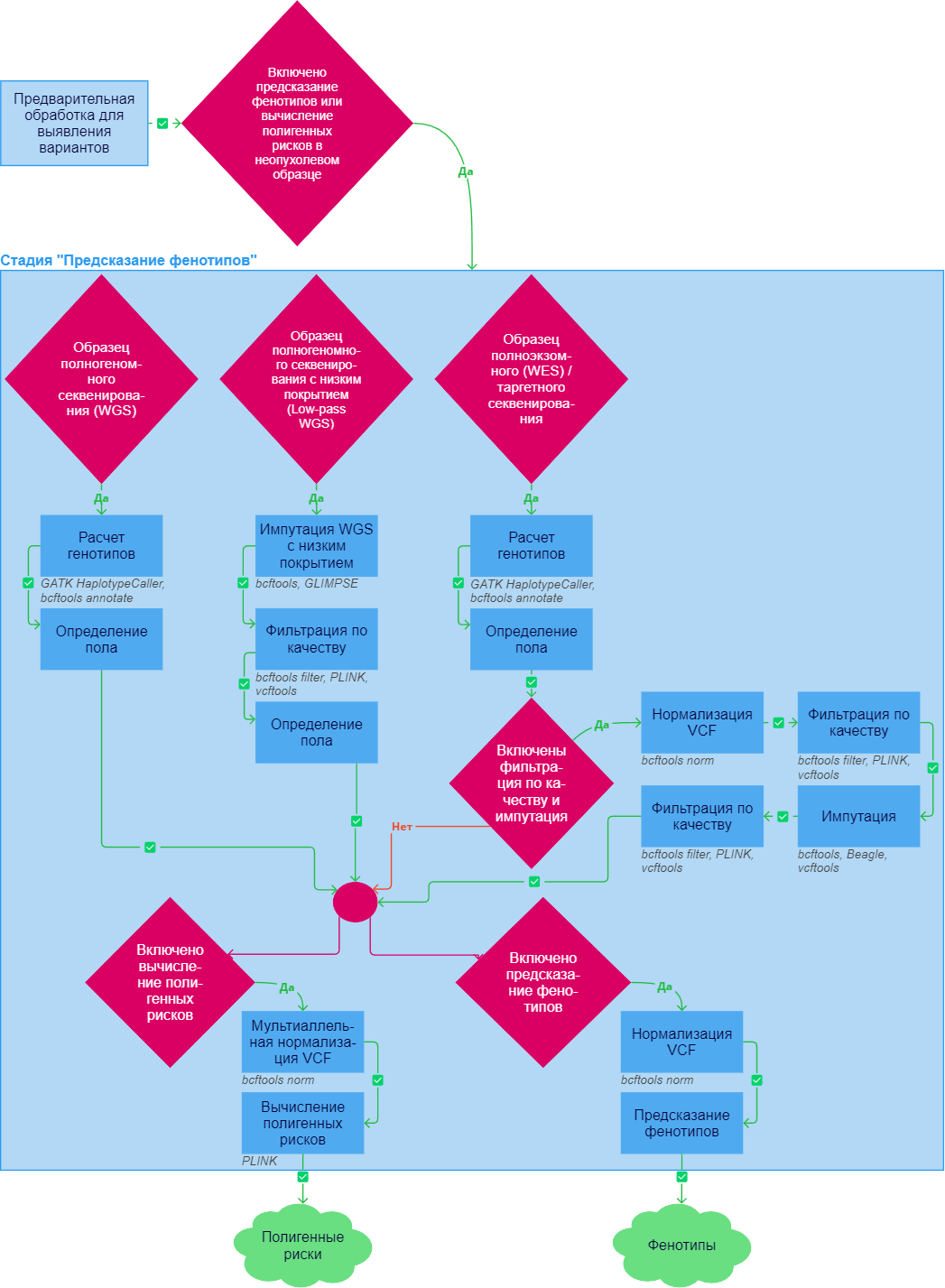
Конвертация BAM в VCF#
Для перехода к задачам предсказания фенотипов или вычисления полигенных рисков необходимо сформировать из файла выравнивания образца в формате BAM файл в формате VCF с герминальными вариантами, удовлетворяющими определённым условиям. Эта предобработка отличается для образцов с разными типами секвенирования.
Предобработка образца полногеномного секвенирования#
В случае определения типа секвенирования образца как "WGS" (whole-genome sequencing; полногеномное секвенирование) конвертация файла выравнивания в VCF файл происходит в ходе задачи "Расчет генотипов".
Расчет генотипов#
GATK HaplotypeCaller распознает в файле выравнивания образца сайты герминальных вариантов и надёжные референсные сайты, которые необходимы для расчёта оценки полигенного риска фенотипов. Распознавание осуществляется посредством локальной сборки гаплотипов de novo в активном регионе. Другими словами, всякий раз, когда программа встречает регион с признаками вариации, она отбрасывает существующую информацию о картировании и полностью пересобирает прочтения в этом регионе. Это повышает точность при распознавании регионов, которые традиционно сложно распознать, например, когда они содержат разные типы вариантов близко друг к другу. Для каждого потенциального сайта варианта программа применяет правило Байеса, используя вероятности аллелей с учетом данных прочтения для расчета вероятности каждого генотипа на образец с учетом данных прочтения, наблюдаемых для этого образца. Затем образцу присваивается наиболее вероятный генотип.
Далее варианты аннотируются идентификторами из базы dbSNP (rsId) с помощью bcftools annotate. Получившийся файл сжимается в GZIP архив с помощью bgzip. Его можно скачать в разделе "Файлы с результатами" в деталях задачи "Расчет генотипов" ("Скачать VCF_GZ"). Кроме того, файл индексируется с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать VCF_TBI").
Предобработка образца полногеномного секвенирования с низким покрытием#
В случае определения типа секвенирования образца как "Low-pass WGS" (полногеномное секвенирование с низким покрытием) конвертация файла выравнивания в VCF файл происходит в ходе задачи "Импутация WGS с низким покрытием", за которой следует обязательная задача "Фильтрация по качеству" (описана далее).
Внимание!
Пайплайн предсказания фенотипов и вычисления полигенных рисков в данных полногеномного секвенирования с низким покрытием является экспериментальным.
Импутация WGS с низким покрытием#
Импутация - это статистический метод восстановления отсутствующих генетических данных на основе анализа гаплотипов в референтной выборке. Сначала происходит распознавание генотипа для данных с низким покрытием с помощью bcftools mpileup и bcftools call. В результате генерируется файл формата VCF, содержащий вероятности генотипа (Genotype Likelihoods; GLs) для каждого сайта из референсной панели. Этот файл далее принимается в качестве входных данных инструментом GLIMPSE. Кроме того, происходит разделение вариантов по хромосомам для параллельной импутации. Затем для более оптимизированного вычисления GLIMPSE chunk разделяет данные на фрагменты для импутации и фазирования и фазирует распространенные варианты. GLIMPSE phase импутирует и фазирует данные секвенирования с низким покрытием. В качестве референсной панели используется панель, созданная на основе коллекции образцов с 30-кратным покрытием из проекта 1000 Genomes Project. GLIMPSE ligate фазирует редкие варианты на скаффолд распространенных вариантов и сшивает все фазированные фрагменты вместе в один файл с целой хромосомой. Наконец, происходит объединение всех хромосом вместе и конвертация в VCF с помощью bcftools concat. Затем этот файл сортируется по хромосомам и сжимается в GZIP архив с помощью bgzip. Получившийся файл с импутированными вариантами в формате VCF можно скачать в разделе "Файлы с результатами" в деталях задачи "Импутация WGS с низким покрытием" ("Скачать VCF_GZ"). Кроме того, файл индексируется с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать VCF_TBI").
Предобработка образца полноэкзомного или таргетного секвенирования#
В случае определения типа секвенирования образца как "Панель" (секвенирование с помощью таргетной панели) или "WES" (whole-exome sequencing; полноэкзомное секвенирование) конвертация файла выравнивания в VCF файл происходит в ходе задачи "Расчет генотипов" (описана выше), за которой следуют задачи контроля качества, если включен соответствующий параметр "Enable QC and imputation". Контроль качества включает задачи "Нормализация VCF", "Фильтрация по качеству", "Импутация" и завершается повторной фильтрацией по качеству.
На заметку
Если в результате одной из задач фильтрации по качеству образец не проходит проверку контроля качества, то выполнение стадии "Геномные предсказания" на этом прерывается, а вычисление олигогенных рисков, вычисление полигенных рисков и/или анализ происхождения не происходит. Анализ образца может продолжиться генерацией отчётов. Вы можете отключить контроль качества и импутацию, однако результаты в этом случае могут оказаться неудовлетворительными.
Нормализация VCF#
На этом этапе осуществляются проверка того, соответствуют ли референсные аллели в файле референсной последовательности; разбивка мультиаллельных сайтов на биаллельные записи; вывод только первой записи для дублирующихся строк с помощью bcftools norm. Получившийся файл сжимается в GZIP архив с помощью bgzip. Его можно скачать в разделе "Файлы с результатами" в деталях задачи "Нормализация VCF" ("Скачать VCF_GZ"). Кроме того, файл индексируется с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать VCF_TBI").
Фильтрация по качеству#
- Исключение из анализа вариантов, у которых нет информации о сайте (например, ./.), с помощью bcftools filter.
- Конвертация файла в формате VCF в бинарный набор файлов, который является способом представления выявленных генотипов, и удаление всех вариантов с одинаковыми идентификаторами, кроме первого встретившегося в файле (варианты без идентификатора пропускаются) с помощью PLINK. Файл с подробным описанием выполнения этой задачи можно скачать в разделе "Файлы с результатами" в деталях задачи "Фильтрация по качеству" ("Скачать convert log TXT").
- Контроль качества - проверка и удаление несоответствующих критериям образцов и однонуклеотидных полиморфизмов (ОНП) с помощью PLINK. Включает предварительные этапы (проверка доли выявления образцов и ОНП и проверка пола) и итерационные этапы (повторяются итеративно до тех пор, пока ошибки не перестанут находиться).
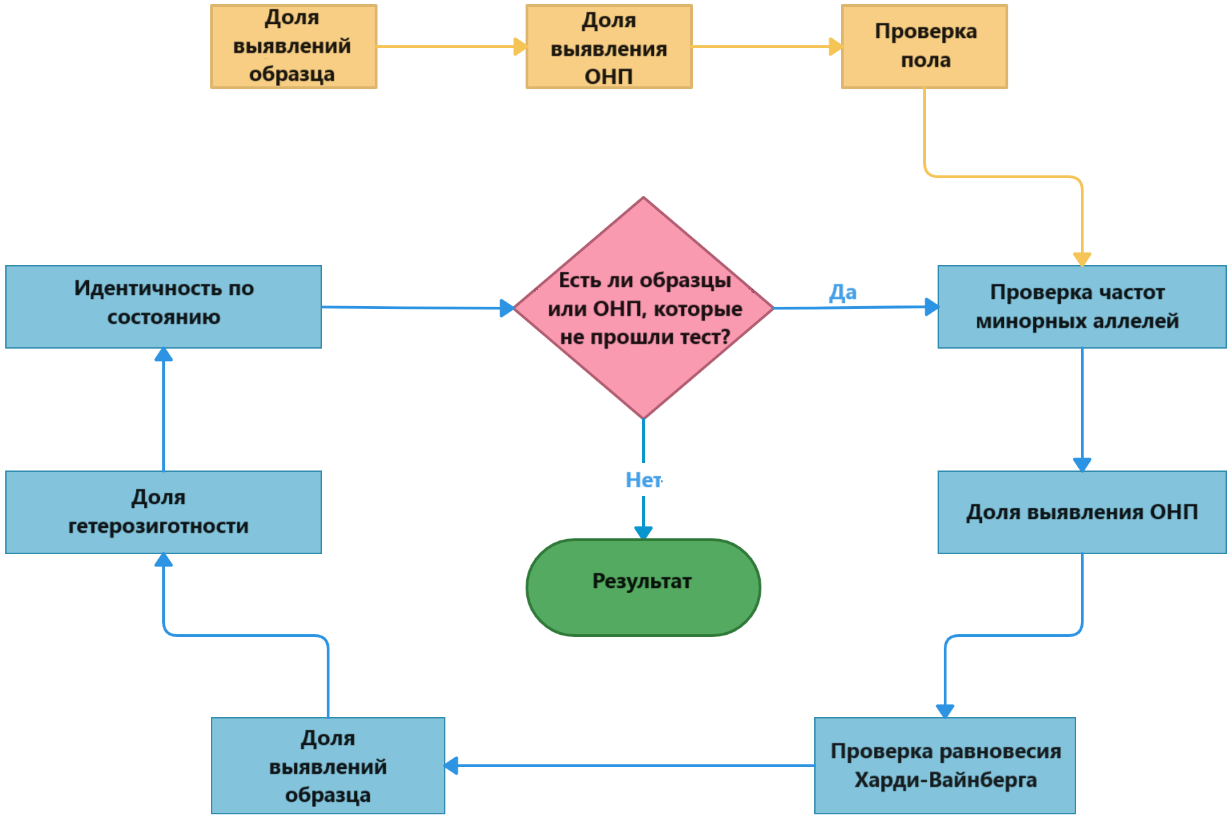
- Проверка данных на наличие образцов с крайне низкой долей выявления. Образцы с высокой долей пропущенных выявлений являются следствием низкого качества ДНК и удаляются из анализа. Порог для доли выявления образцов: > 0.5.
- Проверка данных на наличие однонуклеотидных полиморфизмов (ОНП) с крайне низкой долей выявления. Доля пропущенных выявлений ОНП - это доля образцов, генотипы которых не выявлены для данного ОНП. ОНП с высокой долей отсутствующих генотипов (обычно > 5%) предполагают некоторые проблемы с процессом генотипирования, поэтому такие ОНП исключаются из анализа. Порог для доли выявления ОНП: > 0.5.
- Проверка пола и удаление образцов с неправильным полом. Проверка пола основана на оценке гетерозиготности X-сцепленных ОНП. По умолчанию коэффициент инбридинга X-хромосомы F < 0,2 определяет пол в образцах как женский, а F > 0,8 — как мужской.
- Проверка частоты минорных аллелей (minor allele frequency; MAF). ОНП с MAF < 1% исключаются из последующего анализа, поскольку SNP-чипы, генотипирующие редкие варианты (т.е. локус с MAF < 1%), сложны и подвержены ошибкам. Таким образом, очень низкочастотные аллели, вероятно, являются следствием ошибки генотипирования и могут привести к ложным ассоциациям.
- Фильтрация ОНП по доле выявления с порогом > 0.98.
- Проверка равновесия Харди-Вайнберга. Согласно допущению Харди-Вайнберга, частоты аллелей и генотипов можно оценить от одного поколения к другому. Отмечается, что отклонение от равновесия Харди-Вайнберга может происходить из-за отбора, популяционного смешивания, загадочного родства, ошибки генотипирования и истинной генетической ассоциации. Поэтому для контроля качества проверяется, не отклоняются ли ОНП от равновесия Харди-Вайнберга.
- Фильтрация образцов по доле выявления с порогом > 0.98.
- Гетерозиготность образцов. Доля гетерозиготных генотипов в геноме образца может выявить некоторые проблемы с генотипированием, такие как загрязнение образца и инбридинг. Из анализа удаляются образцы, которые отклоняются на ± 3 SD (стандартное отклонение) от средней гетерозиготности образца.
- Идентичность по проверке состояния. Высокая степень родства между образцами может привести к
усилению ассоциации. Чтобы исследовать загадочное родство, мы рассчитываем матрицу родства и фильтруем
образцы с близкими отношениями. Порог идентичности по состоянию: < 0.0925.
Файл с подробным описанием выполнения задачи контроля качества можно скачать в разделе "Файлы с результатами" в деталях задачи "Фильтрация по качеству" ("Скачать QC log TXT").
- Повторная проверка и фильтрация вариантов и генерация нового бинарного набор файлов с отфильтрованными
образцами и ОНП с помощью PLINK.
Файл с подробным описанием выполнения этой задачи можно скачать в разделе "Файлы с результатами" в деталях задачи "Фильтрация по качеству" ("Скачать merge log TXT").
Файл с вариантами, удаленными из анализа, можно скачать в том же разделе ("Скачать Skipped variants TXT"). Для каждого варианта указаны причина удаления и номер итерации контроля качества, на которой произошло удаление.
Полный отчёт о контроле качества данных можно открыть в том же разделе ("Открыть QC report HTML").
Файл с образцами, удаленными из анализа, можно скачать в том же разделе ("Скачать Removed samples TXT"). Для каждого образца указаны причина удаления и номер итерации контроля качества, на которой произошло удаление. - Исключение из анализа вариантов, не прошедших фильтрацию, с помощью vcftools.
- Сжатие файла в GZIP архив с помощью bgzip. Получившийся файл с отфильтрованными вариантами в формате VCF можно скачать в разделе "Файлы с результатами" в деталях задачи "Фильтрация по качеству" ("Скачать Filtered VCF_GZ").
- Индексация файла с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать Filtered VCF_TBI").
Импутация#
Импутация - это статистический метод восстановления отсутствующих генетических данных на основе анализа гаплотипов в референтной выборке.
- Индексация файла VCF с помощью bcftools index.
- Разделение вариантов по хромосомам с помощью bcftools view для параллельной импутации.
- Определение генотипов и импутация негенотипированных маркеров с
помощью Beagle.
- Объединение импутированных вариантов, разбитых по хромосомам, в один файл в формате VCF с помощью bcftools concat.
- Индексация файла VCF с помощью bcftools index.
- Сравнение исходного файла и файла, полученного после импутации, и получение файла с неимпутированными вариантами с помощью vcftools.
- Сжатие файла с неимпутированными вариантами в GZIP архив с помощью bgzip.
- Индексация файла с неимпутированными вариантами с помощью bcftools index.
- Фильтрация импутированных вариантов по порогу DR2 (dosage R-squared) > 0.3 с помощью bcftools filter.
- Индексация файла с импутированными и отфильтрованными вариантами с помощью bcftools index.
- Объединение неимпутированных и отфильтрованных импутированных вариантов в один файл в формате VCF с помощью bcftools concat.
- Индексация объединенного файла с
помощью bcftools index.
Файл с подробным описанием выполнения задачи импутации можно скачать в разделе "Файлы с результатами" в деталях задачи "Импутация" ("Скачать Impute log TXT").
Файл с неимпутированными и отфильтрованными импутированными вариантами в формате VCF можно скачать в том же разделе ("Скачать Imputed VCF_GZ").
Индексный файл к VCF файлу можно скачать в том же разделе ("Скачать Imputed VCF_TBI").
Определение пола#
Независимо от типа секвенирования, для всех образцов импутируется информация о поле из данных об уровне гомозиготности X-хромосомы. Коэффициент инбридинга X-хромосомы F рассчитывается по следующей формуле:
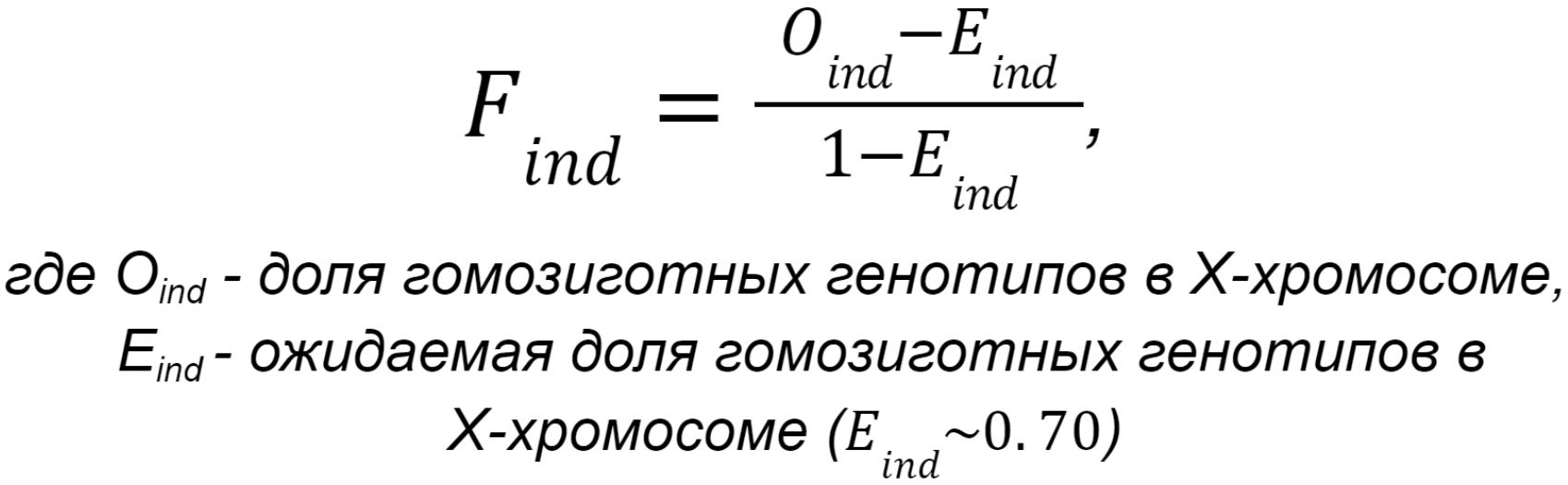
Если F < 0.2, то пол определяется как женский. Если F > 0.8, то пол определяется как мужской. Если 0.2 < F < 0.8, то пол невозможно определить однозначно.
Пол пациента используется для формирования отчёта по полигенным признакам. Если пользователь указал пол вручную, используется именно это значение; в противном случае применяется пол, определённый по генетическим данным.
Вычисление полигенных рисков#
Вычисление полигенных рисков запускается после успешно пройденных шагов предобработки образца, если включен соответствующий параметр.
1. Мультиаллельная нормализация VCF#
Мультиаллельная нормализация VCF - необходимый этап перед вычислением полигенных рисков. Включает выравнивание по левому краю и нормализация инделов; проверку того, соответствуют ли референсные аллели в файле референсной последовательности; объединение биаллельных сайтов в мультиаллельные записи, которые выполняются с помощью bcftools norm. Получившийся файл с нормализованными вариантами сжимается в GZIP архив с помощью bgzip. Его можно скачать в разделе "Файлы с результатами" в деталях задачи "Мультиаллельная нормализация VCF" ("Скачать VCF_GZ"). Кроме того, файл индексируется с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать VCF_TBI").
2. Вычисление полигенных рисков#
Вычисление полигенных рисков заключается в применении системы линейной оценки к каждой выборке с помощью PLINK. Варианты без информации о сайте (генотип ./. и подобные), без идентификатора или с несовпадающими кодами аллелей не учитываются в анализе. Генетические данные пациента должны включать варианты, представленные в моделе полигенных рисков, за исключением небольшой их доли, устанавливаемой пороговым значением. Вычисление полигенных рисков считается возможным, если генетические данные пользователя содержат не менее 95% всех вариантов, которые представлены в модели.
Оценка полигенного риска (polygenic risk score; PRS) - число, которое суммирует предполагаемый размер эффектов многих однонуклеотидных полиморфизмов (ОНП) на фенотип человека. Для каждого признака значение полигенного риска вычисляется по следующей формуле:
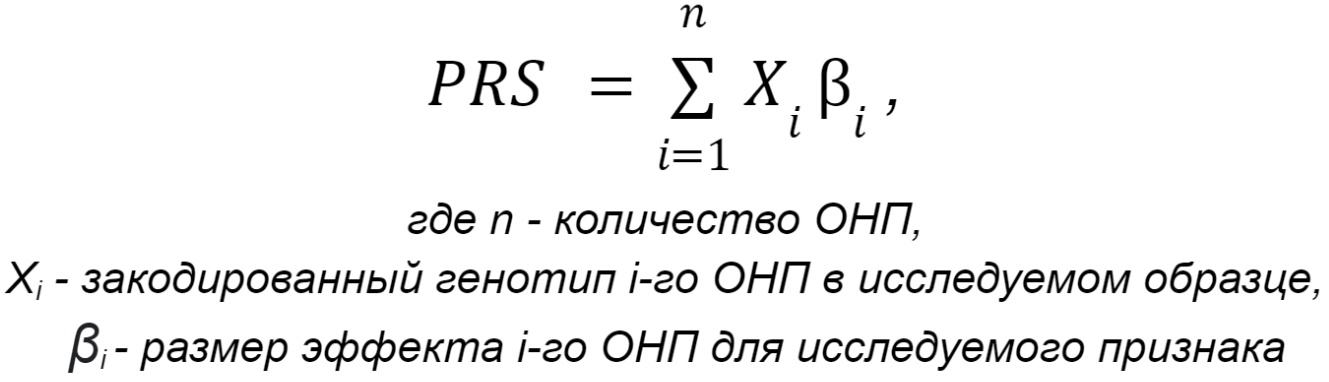
Генотипы закодированы следующим образом: пусть, аллель A - эффектный, а аллель G - неэффектный. Тогда
численный код генотипа AA - 2, генотипа AG - 1, а генотипа GG - 0.
Предполагаемые размеры эффектов ОНП рассчитываются на основе данных полногеномного поиска ассоциаций
(genome-wide association study; GWAS), который позволяет сопоставить фенотипические признаки с геномными
вариантами в человеческих популяциях.
Оценка полигенного риска отражает предполагаемую генетическую предрасположенность человека к исследуемому
признаку и может использоваться в качестве предиктора этого признака в предсказательной модели. Другими словами,
PRS оценивает, насколько вероятно, что человек будет иметь исследуемый признак, только на основе генетических
данных и без учёта факторов окружающей среды.
Вычисляемые полигенные риски:
- Рост;
- Масса тела;
- Индекс массы тела (ИМТ; Body mass index; BMI);
- Жировая масса туловища;
- Безжировая масса туловища;
- Окружность бедер;
- Предрасположенность к избыточному весу;
- Уровень инсулиноподобного фактора роста 1;
- Уровень глюкозы натощак;
- Уровень инсулина натощак;
- Уровень гемоглобина;
- Гематокрит;
- Минеральная плотность кости;
- Предрасположенность к ишемической болезни сердца;
- Предрасположенность к воспалительному заболеванию кишечника;
- Предрасположенность к сахарному диабету 2-го типа;
- Предрасположенность к хронической почечной недостаточности;
- Предрасположенность к астме;
- Предрасположенность к идиопатическому легочному фиброзу;
- Предрасположенность к гипертиреозу;
- Предрасположенность к алкоголь-ассоциированному циррозу печени;
- Предрасположенность к болезни Альцгеймера;
- Предрасположенность к болезни Паркинсона;
- Предрасположенность к раку предстательной железы;
- Предрасположенность к раку молочной железы;
- Предрасположенность к колоректальному раку;
- Показатель невротичности;
- Склонность к принятию риска;
- Риск госпитализации при COVID-19;
- Риск тяжелого течения COVID-19.
Для каждого риска в результате выполнения задачи формируются три файла, которые можно скачать в разделе "Файлы с результатами" в деталях задачи "Вычисление полигенных рисков":
- Файл с подробным описанием вычисления риска - "Скачать [название риска] Prs log TXT".
- Файл с суммарной оценкой риска для этого образца - "Скачать [название риска] Score TSV". Также этот файл можно открыть в таблицах Google.
- Файл со списком идентификаторов вариантов, использованных для вычисления риска, - "Скачать [название риска] Used Variants TSV". Также этот файл можно открыть в таблицах Google.
Вычисление олигогенных рисков#
Вычисление олигогенных рисков запускается после успешно пройденных шагов предобработки образца, если включен соответствующий параметр.
1. Нормализация VCF#
Вначале VCF файл нормализуется путём выравнивания по левому краю и нормализации инделов; проверки того, соответствуют ли референсные аллели в файле референсной последовательности; разбивки мультиаллельных сайтов на биаллельные записи; вывода только первой записи для дублирующихся строк с помощью bcftools norm. Получившийся файл сжимается в GZIP архив с помощью bgzip. Его можно скачать в разделе "Файлы с результатами" в деталях задачи "Нормализация VCF" ("Скачать VCF_GZ"). Кроме того, файл индексируется с помощью tabix. Получившийся индексный файл можно скачать в том же разделе ("Скачать VCF_TBI").
2. Вычисление олигогенных рисков#
Вычисление олигогенных рисков происходит по однонуклеотидным полиморфизмам (single nucleotide polymorphism; SNP), ассоциированным с определенным проявлением признака согласно моделям, основанным на мультиномиальной логистической регрессии (multinomial logistic regression; MLR).
Вычисляемые олигогенные риски:
- Цвет волос;
- Цвет глаз;
- Цвет кожи;
- Веснушчатость;
- Непереносимость лактозы;
- Горький вкус;
- Группа крови;
- Метаболизм алкоголя;
- Тип ушной серы;
- Подмышечный осмидроз;
- Мизофония;
- Движение во время сна;
- Световой рефлекс чихания.
Подробно каждый признак и соответствующая модель предсказания описаны в разделе, посвящённом отчёту по олигогенным признакам.
Получившийся файл с предсказанными олигогенными признаками можно скачать в разделе "Файлы с результатами" в деталях задачи "Вычисление олигогенных рисков" ("Скачать Classifier results JSON").
После стадии "Геномные предсказания" анализ может продолжиться генерацией отчётов.