Загрузка, идентификация и проверка
В случае образца секвенирования одиночных прочтений анализироваться будет один файл и начальная стадия анализа будет называться "Загрузка, идентификация и проверка". В случае образца секвенирования парных прочтений анализироваться будут два парных файла и начальных стадий анализа будет две: "Загрузка, идентификация и проверка первичного файла" и "Загрузка, идентификация и проверка парного файла".
На начальной стадии анализа файл или файлы образца в формате FASTQ или BAM загружаются, определяется их формат и проводится их проверка. При ошибке выполнения любой из перечисленных ниже задач анализ образца останавливается.
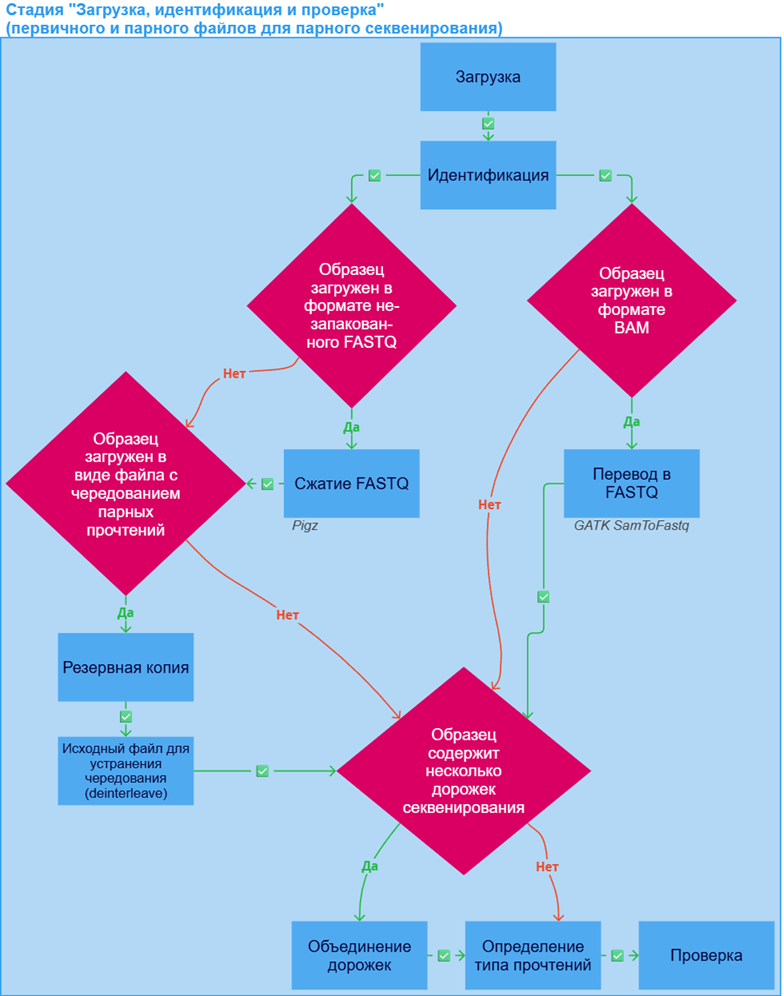
Стадия анализа образца "Загрузка, идентификация и проверка" может включать следующие задачи:
- Загрузка. Если вы загружаете файл(ы) образца с компьютера, а не по ссылке, то загрузка может прерваться. Чтобы её восстановить, воспользуйтесь формой возобновления загрузки.
- Идентификация: определение формата данных и типа секвенатора. Для образца, загруженного в формате BAM, будет произведена проверка на целостность с помощью samtools quickcheck.
- Сжатие FASTQ в GZIP архив, если образец был загружен в виде файла в формате FASTQ, не запакованного в архив или запакованного в архив, отличный от GZIP (например, ZIP, BZIP2, 7-ZIP, XZ, WIM, RAR). Сжатие осуществляется с помощью Pigz.
- Перевод в FASTQ, если образец был загружен в формате BAM. Осуществляется с помощью GATK SamToFastq. Полученные файлы в формате FASTQ сжимаются в GZIP архив с помощью Pigz. Оригинальный файл в формате BAM можно скачать в разделе "Файлы с результатами" в деталях задачи "Перевести в FASTQ" ("Скачать Original BAM").
- Образец может быть загружен в виде файла с чередованием парных прочтений (interleaved). В таком файле для каждой пары прочтений первичное прочтение всегда записывается непосредственно перед соответствующим парным прочтением. Такой образец проходит дополнительные задачи подготовки к анализу Резервная копия и Исходный файл для устранения чередования (deinterleave), необходимые для аккуратного разделения прочтений из файла на первичные и парные и записи их в два отдельных файла. Все задачи до разделения, кроме идентификации, будут записаны в детали стадии "Загрузка, идентификация и проверка первичного файла", а стадия "Загрузка, идентификация и проверка парного файла" будет включать только задачи "Идентификация", "Определение типа прочтений" и "Проверка".
- Объединение дорожек - процесс объединения нескольких файлов FASTQ в один образец в случае, если на этапе загрузки он был распознан или указан вручную как образец секвенирования, представленный несколькими дорожками одной проточной ячейки.
- Определение типа прочтений - экспериментальная функция,
не влияющая на дальнейший ход анализа. В рамках задачи используется встроенный классификатор, основанный
на модели LSTM (long short-term memory; долгая краткосрочная память), который
предсказывает тип нуклеиновой кислоты (ДНК, РНК или UNKNOWN), чья последовательность была отсеквенирована при
получении образца. Результат отображается на странице образца, где при
необходимости его можно изменить вручную.
Задача "Определение типа прочтений" не выполняется, если при добавлении образца была выбрана предустановка настроек с заранее заданным типом прочтений (ДНК или РНК). В этом случае задача включается в пайплайн, но не запускается, а указанный тип устанавливается без дополнительной проверки. - Проверка файла на целостность; проверка того, что на каждое прочтение приходится по четыре строки; проверка того, что длины последовательности и качества последовательности одинаковы.
Загруженный сжатый файл (или файлы) образца в формате FASTQ можно скачать наверху вкладки "Детали процесса анализа". В случае загрузки образца с чередованием парных прочтений для скачивания будут доступны первичный и парный файлы после разделения.
После успешного выполнения стадии "Загрузка, идентификация и проверка" анализ продолжается проверкой качества.