Выравнивание
После успешного выполнения стадии "Проверка качества и очистка" для дальнейшего анализа образца (поиска генетических вариантов, структурных вариаций и вариации числа копий, а также предсказания фенотипов) необходимо выполнить выравнивание (картирование) прочтений на референсный геном. Для выравнивания используется наиболее современная версия генома человека - GRCh38.p14. Если выравнивание исключено из анализа, то после успешного выполнения стадий "Загрузка, идентификация и проверка" и "Проверка качества и очистка" анализ завершается. При ошибке выполнения любой из перечисленных ниже задач анализ образца останавливается.
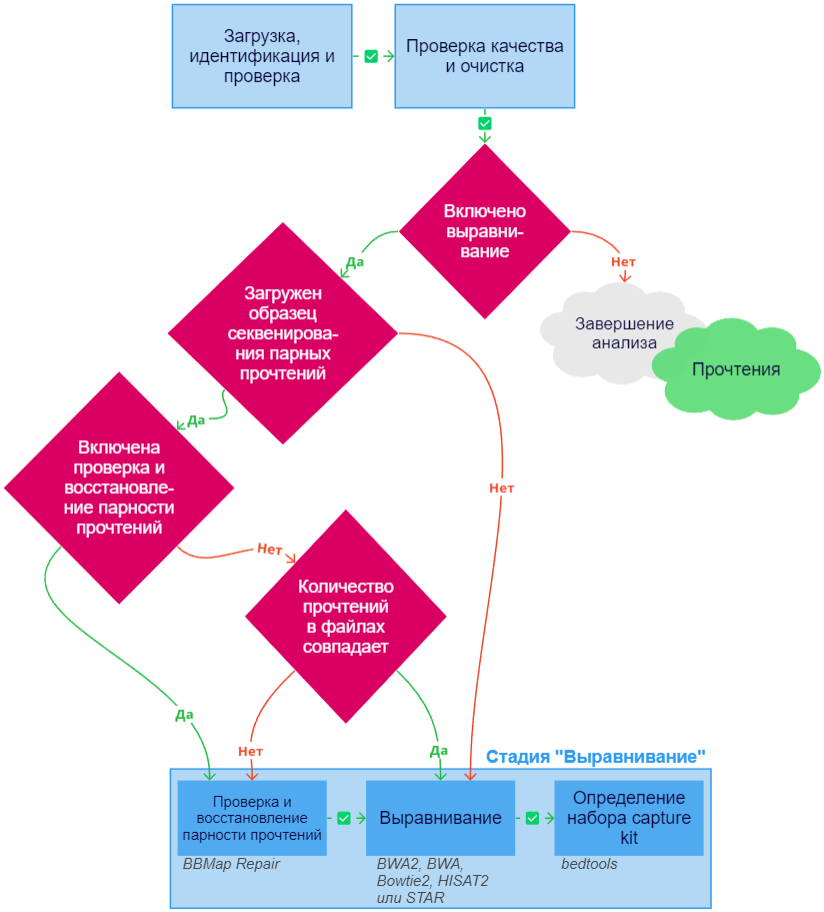
Стадия анализа образца "Выравнивание" может включать следующие задачи:
- Проверка и восстановление парности прочтений запускается, если выполняются следующие условия:
- Анализируется образец парного секвенирования (секвенирование, при котором фрагменты ДНК секвенируют с каждого конца навстречу друг другу);
- Включены проверка и восстановление парности прочтений и/или количество прочтений в парных файлах образца не совпадает.
Проверка и восстановление парности прочтений производится BBMap Repair. В ходе выполнения задачи происходит следующее:
- Сортировка строк в файлах.
- Восстановление порядка в парных файлах: первое прочтение в первом файле должно соответствовать первому прочтению во втором файле и т.д. Порядок восстанавливается за счёт анализа названий прочтений, которые должны быть либо в формате Illumina (идентичный префикс, сопровождаемый "1:" и "2:" или "/1" и "/2"), либо полностью идентичны для обоих прочтений в паре.
- Восстановление парности прочтений, необходимое для выравнивания, чтобы каждое прочтение из одного файла содержало пару во втором. При очистке прочтений парность может быть нарушена, что приводит к образованию синглтонов - прочтений, которые лишились пары в парном файле. По умолчанию синглтоны удаляются из анализа, но их можно оставить в анализе для выравнивания на геном, если включить соответствующий параметр.
- Выравнивание - определение местоположения прочтений образца на референсном геноме версии hg38. По умолчанию выравнивание производится с помощью рекомендуемого сообществом инструмента BWA2 Burrows-Wheeler Aligner (BWA-MEM2) - новейшей и более быстрой версии алгоритма BWA-MEM, который основан на обратном поиске с преобразованием Берроуза-Уилера (BWT) и эффективно и быстро выравнивает короткие прочтения секвенирования на геном человека, допуская мисмэтчи и гэпы1. Этот выравниватель можно поменять в параметрах на BWA Burrows-Wheeler Aligner - более раннюю версию алгоритма BWA-MEM2, Bowtie23, HISAT24 или STAR5. Выравнивание записывается в файл в формате SAM (текстовый файл для хранения биологических последовательностей, выровненных на референсную последовательность).
После выравнивания картированные прочтения сортируются по крайним левым координатам с помощью samtools sort и записываются в файл в формате BAM (бинарный эквивалент формата SAM; занимает меньше места и позволяет быстрее работать с информацией, чем SAM). Получившийся файл можно скачать в разделе "Файлы с результатами" в деталях задачи "Выравнивание" ("Скачать BAM"). Также этот файл можно открыть в IGV, нажав на ссылку "Открыть в IGV браузере". Полученный BAM файл индексируется с помощью samtools index. Получившийся индексный файл можно скачать в том же разделе ("Скачать BAI").
На заметку
Если вы хотите добавить в вашу десктопную версию IGV трек выравнивания, полученного в результате анализа загруженного вами образца в Genomenal, вы можете сделать это через ссылку. Для этого сделайте следующее:
- Нажмите правой кнопкой мыши на ссылку на файл выравнивания "Скачать BAM" и выберите опцию "Копировать адрес ссылки".
- Загрузите трек через URL в вашу десктопную версию IGV, как это описано здесь.
- Нажмите правой кнопкой мыши на ссылку на индексный файл выравнивания "Скачать BAI" и выберите опцию "Копировать адрес ссылки".
- Добавьте URL индексного файла в соответствующее поле в IGV.
- Нажмите "OK". Готово! Трек выравнивания образца добавлен в IGV.
В разделе "Метрики" в деталях задачи "Выравнивание" приведены метрики оценки качества выравнивания:

Для каждой метрики приведены:
- название метрики;
- описание результата проверки соответствия определенного показателя выравнивания образца и порога этой метрики: приведены значение показателя для образца (например, доля картированных прочтений в файле выравнивания - Mapped reads) и использованный порог метрики, при котором файл выравнивания считается качественным (можно поменять в параметрах);
- результат оценки качества выравнивания
по метрике:
 , если выравнивание образца удовлетворяет порогу метрики,
, если выравнивание образца удовлетворяет порогу метрики,
или ,
если не удовлетворяет.
,
если не удовлетворяет.
Метрики оценки качества выравнивания#
| Метрика | Значение порога метрики, при котором файл выравнивания считается качественным (значение по умолчанию, может быть изменено в параметрах) |
| Mapped reads (картированные прочтения) | В файле выравнивания не менее 85% картированных прочтений. |
| Multiple alignments (множественные выравнивания) | В файле выравнивания не более 15% множественных выравниваний одного и тоже прочтения на геном. |
| Forward/reverse balance (равновесие прочтений с прямой и обратной цепи) | Разница в количестве прочтений с прямой цепи и прочтений с обратной цепи не более 10% в файле выравнивания. |
| Paired mapped reads (картированные парные прочтения) | В файле выравнивания не менее 80% картированных парных прочтений. |
| Paired properly mapped reads (правильно картированные парные прочтения) | В файле выравнивания не менее 75% правильно картированных парных прочтений. |
| Coverage per nucleotide (понуклеотидное покрытие) | Минимально допустимое понуклеотидное покрытие на геноме - 0.1. |
Если значение метрики в файле выравнивания образца не удовлетворяет установленному порогу, то файл отмечается как не удовлетворяющий требованиям метрики, т.е. выравнивание завершается успешно, а в статусе стадии указывается количество метрик, которые не соответствуют критериям. Если же никакие прочтения образца не выровнялись на геном, то стадия "Выравнивание" завершается с ошибкой и анализ останавливается.
На заметку
Если вы анализируете данные ампликоновых панелей, рекомендуем настроить параметры выравнивания для фильтрации неспецифических выравниваний, как описано здесь.
Если в анализ образца включена хотя бы одна из стадий: выявление соматических SNVs/Indels, герминальных SNVs/Indels, структурных вариаций, вариации числа копий, вычисление олигогенных рисков, вычисление полигенных рисков, pасчет фармакогенетики или анализ происхождения, то после успешного выполнения задачи "Выравнивание" начнётся выполнение стадии "Предварительная обработка для выявления вариантов".
- Определение набора capture kit
Если пользователь определил тип секвенирования образца как WGS (whole-genome sequencing; полногеномное секвенирование), то определение набора capture kit не проводится, а тип секвенирования может остаться WGS или переопределиться в Low-pass WGS (полногеномное секвенирование с низким покрытием). WGS и Low-pass WGS разделяются по среднему покрытию образца: тип секвенирования образцов со средним покрытием меньше 4x определяется как Low-pass WGS, а выше или равно 4x - как WGS.
Если пользователь определил тип секвенирования как Направленный отбор, то тип секвенирования может определиться как Панель (секвенирование с помощью таргетной панели) или WES (whole-exome sequencing; полноэкзомное секвенирование). Далее определяется наиболее подходящий для образца набор capture kit (таргетная панель, используемая при направленном отборе) или вычисляются статистики покрытия образца набором capture kit, выбранным пользователем:
- bedtools genomecov вычисляет покрытие выровненных последовательностей образца на референсный геном и выдает выходные данные о полногеномном покрытии в формате bedGraph (Файл A).
- bedtools intersect выявляет совпадения между Файлом A и либо набором capture kit, выбранным для образца пользователем, либо каждым набором capture kit, который встроен или загружен в систему, если пользователь не выбрал capture kit. Для этих capture kit рассчитываются индекс покрытия, глубина, суммарная длина интервалов и длина пересечения покрытия выровненных последовательностей образца с интервалами capture kit, а также процент покрытия.
- Для capture kit, выбранного пользователем, или для наиболее подходящего набора capture kit среди рассмотренных на предыдущем этапе наборов bedtools complement строит файл со всеми интервалами в геноме, которые не покрыты хотя бы одним интервалом в файле capture kit (Файл B).
- bedtools intersect находит совпадения между Файлом A и Файлом B, вычисляя индекс покрытия, глубину и длину пересечения этих файлов.
В результате, если пользователь не выбрал capture kit и если в ходе выполнения задачи удалось найти наиболее подходящий для образца набор capture kit, то анализ образца идёт дальше с этим набором. Если подходящий capture kit не удалось найти, то для образца не будут выполнены фильтрация SNVs/Indels (поиск вариантов в тех же геномных интервалах, в которых проводилось таргетного секвенирование) и выявление вариации числа копий. Файл со статистиками набора capture kit, выбранного пользователем или определенного системой, можно скачать в разделе "Файлы с результатами" в деталях задачи "Определение набора capture kit" ("Скачать Capture kit stats JSON").
- Vasimuddin M., Sanchit M., Heng L., Srinivas A. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. IEEE Parallel and Distributed Processing Symposium (IPDPS) (2019)↩
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 (2013)↩
- Langmead B., Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357:359 (2012)↩
- Kim D., Paggi J.M., Park C. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907:915 (2019)↩
- Dobin A., Davis C.A., Schlesinger F. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15:21 (2013)↩