Предварительная обработка для выявления вариантов
После успешного выполнения задачи "Выравнивание", входящей в одноименную стадию, для наиболее точного выявления вариантов результаты выравнивания рекомендуется дополнительно обработать. Это повышает чувствительность к низкочастотным вариантам, способствует более точному подсчету количества копий, который зависит от количества прочтений, а также ускоряет обработку образца. Предварительная обработка для выявления вариантов запускается, если хотя бы одна из следующих стадий включена в анализ образца: выявление соматических SNVs/Indels, герминальных SNVs/Indels, структурных вариаций, вариации числа копий, вычисление олигогенных рисков, вычисление полигенных рисков, расчет фармакогенетики или анализ происхождения. При ошибке выполнения любой из перечисленных ниже задач анализ образца останавливается.
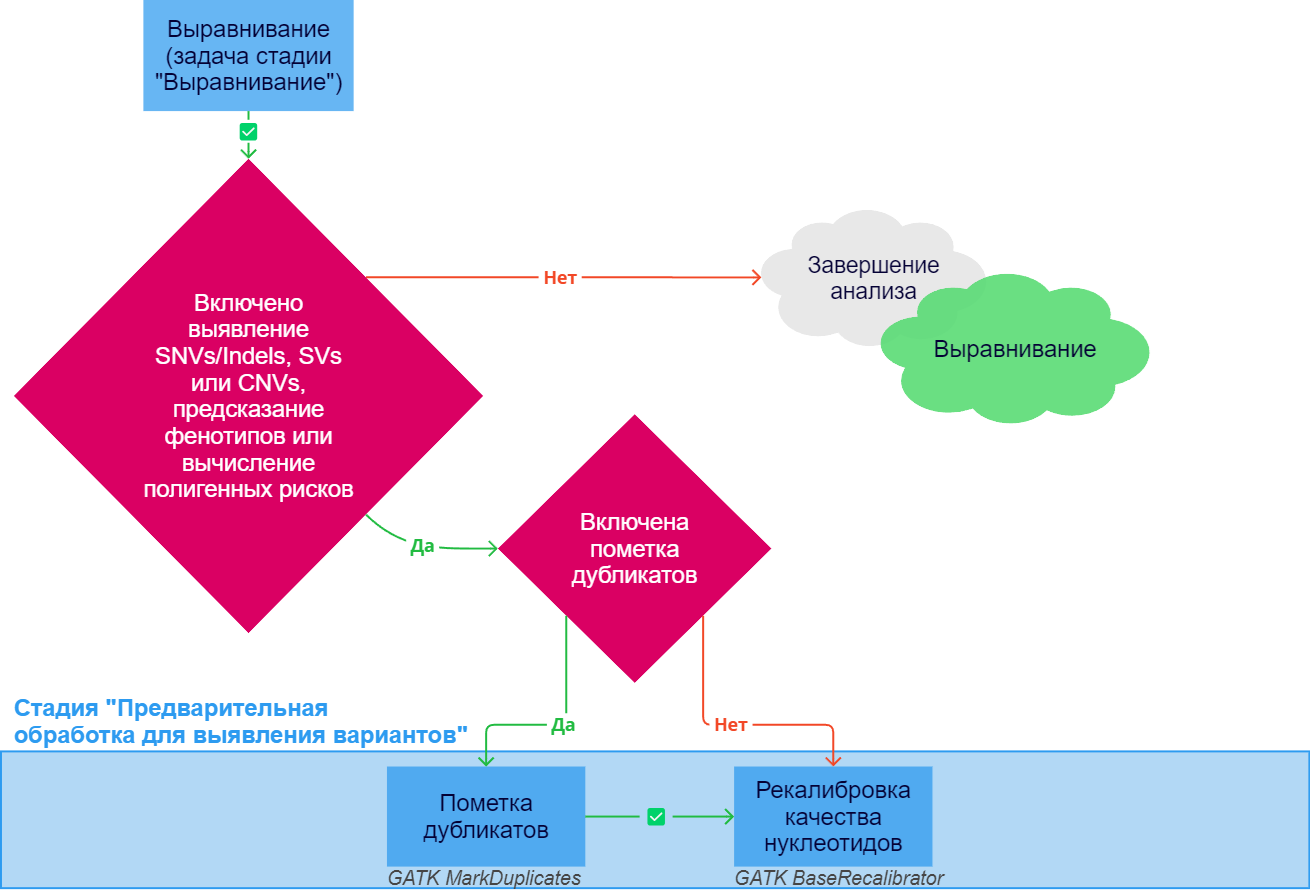
Стадия анализа образца "Предварительная обработка для выявления вариантов" может включать следующие задачи:
Пометка дубликатов, если для образца включен соответствующий параметр. В процессе секвенирования количество оригинальных последовательностей фрагментированной ДНК увеличивается в полимеразной цепной реакции (ПЦР). Для дальнейшего анализа очень важно учесть дублирующиеся прочтения одного и того же фрагмента. Если вы хотите сохранить все прочтения для дальнейшего анализа (например, если вы работаете с ампликонами), то вы можете отключить пометку дубликатов.
Пометка дубликатов выполняется с помощью инструмента GATK MarkDuplicates, который находит в файле выравнивания повторяющиеся прочтения, определяет, какие из прочтений происходят от оригинальных последовательностей, а какие являются копиями (дубликатами), и помечает дубликаты. Дальнейший анализ ведется только по оригинальным прочтениям. Итоговый файл в формате BAM сортируется по координатам и для него создается индексный файл. Кроме того, в ходе выполнения задачи создается файл с метриками, в котором указывается количество дубликатов как для одиночных, так и для парных прочтений. Этот файл можно скачать в разделе "Файлы с результатами" в деталях задачи "Пометка дубликатов" ("Скачать duplication metrics TXT").Рекалибровка качества нуклеотидов. Секвенатор присваивает каждому нуклеотиду качество, которое представляет собой логарифм вероятности того, что при секвенировании этот нуклеотид был определен верно, но многие секвенаторы имеют тенденцию к недооценке качества секвенирования и к другим ошибкам. Рекалибровка качества нуклеотидов позволяет исправить систематическое отклонение, влияющее на присвоение секвенатором качества нуклеотидам, что уменьшает ошибку и улучшает качество выявления вариантов в дальнейшем.
Рекалибровка качества нуклеотидов (Base Quality Score Recalibration; BQSR) выполняется в три этапа:
- Эмпирический расчёт ошибки и поиск закономерностей того, как ошибка изменяется в зависимости от особенностей определения нуклеотидов по всем нуклеотидам, с помощью GATK BaseRecalibrator. Соответствующие наблюдения записываются в таблицу рекалибровки на основе таких ковариат, как группа прочтения, заявленная оценка качества, машинный цикл и нуклеотидный контекст. Работает только в определенных интервалах с известными полиморфными сайтами, которые используются для исключения из анализа областей вокруг известных полиморфизмов. Все найденные несоответствия с референсной последовательностью рассматриваются как ошибки и указывают на низкое качество нуклеотидов. Затем вычисляется эмпирическая вероятность ошибки (p) с учётом конкретных ковариат, наблюдаемых в этом сайте: p = количество несоответствий / количество наблюдений. Выходной файл представляет собой таблицу, которая содержит несколько значений ковариат, количество наблюдений, количество несоответствий и эмпирический показатель качества. Таблицу рекалибровки можно скачать в разделе "Файлы с результатами" в деталях задачи "Рекалибровка качества нуклеотидов" ("Скачать recalibration table TXT").
- Применение числовых поправок к каждому отдельному определению нуклеотида (рекалибровка качества нуклеотидов прочтений образца) в пределах определенных интервалов на основе закономерностей, выявленных на первом этапе и записанных в таблице рекалибровки, и запись рекалиброванных данных в новый файл в формате BAM с помощью GATK ApplyBQSR.
- Объединение файлов BAM с результатами рекалибровки в пределах определенных интервалов в один с помощью GATK MergeSamFiles. Итоговый файл в формате BAM сортируется по координатам и для него создается индексный файл.
Итоговый файл выравнивания образца после предварительной обработки можно скачать в разделе "Файлы с результатами" в деталях задачи "Рекалибровка качества нуклеотидов" ("Скачать BAM"). Также этот файл можно открыть в IGV, нажав на ссылку "Открыть в IGV браузере". Индексный файл к итоговому файлу BAM можно скачать там же ("Скачать BAI").
На заметку
Если вы хотите добавить в вашу десктопную версию IGV трек выравнивания образца после предварительной обработки, полученного с помощью Genomenal, вы можете сделать это через ссылку. Для этого сделайте следующее:
- Нажмите правой кнопкой мыши на ссылку на файл выравнивания "Скачать BAM" и выберите опцию "Копировать адрес ссылки".
- Загрузите трек через URL в вашу десктопную версию IGV, как это описано здесь.
- Нажмите правой кнопкой мыши на ссылку на индексный файл выравнивания "Скачать BAI" и выберите опцию "Копировать адрес ссылки".
- Добавьте URL индексного файла в соответствующее поле в IGV.
- Нажмите "OK". Готово! Трек выравнивания образца после предварительной обработки добавлен в IGV.
После успешного выполнения стадии "Предварительная обработка для выявления вариантов" анализ продолжается вычислением покрытия, а также следующими стадиями, если они включены в анализ: "Выявление соматических SNVs/Indels" (для образца опухолевой ткани), "Выявление герминальных SNVs/Indels" (для одиночного образца опухолевой ткани или образца неопухолевой ткани), "Выявление структурных вариаций", "Выявление вариации числа копий" и/или "Геномные предсказания" (для образца неопухолевой ткани).