Выявление вариации числа копий
После успешного выполнения стадии "Предварительная обработка для выявления вариантов" для образца может запуститься выявление вариации числа копий (copy number variations, CNVs), во время которого определяется, сколько копий участка ДНК было в среднем представлено в секвенированном геноме. Для запуска стадии необходимо, чтобы она была включена в анализ и чтобы для образца были определены тип секвенирования и набор реагентов capture kit (автоматически или пользователем вручную). Пайплайн выявления вариации числа копий различается для анализа неопухолевых и одиночных опухолевых образцов и анализа опухолевых образцов из набора опухоль/контроль.
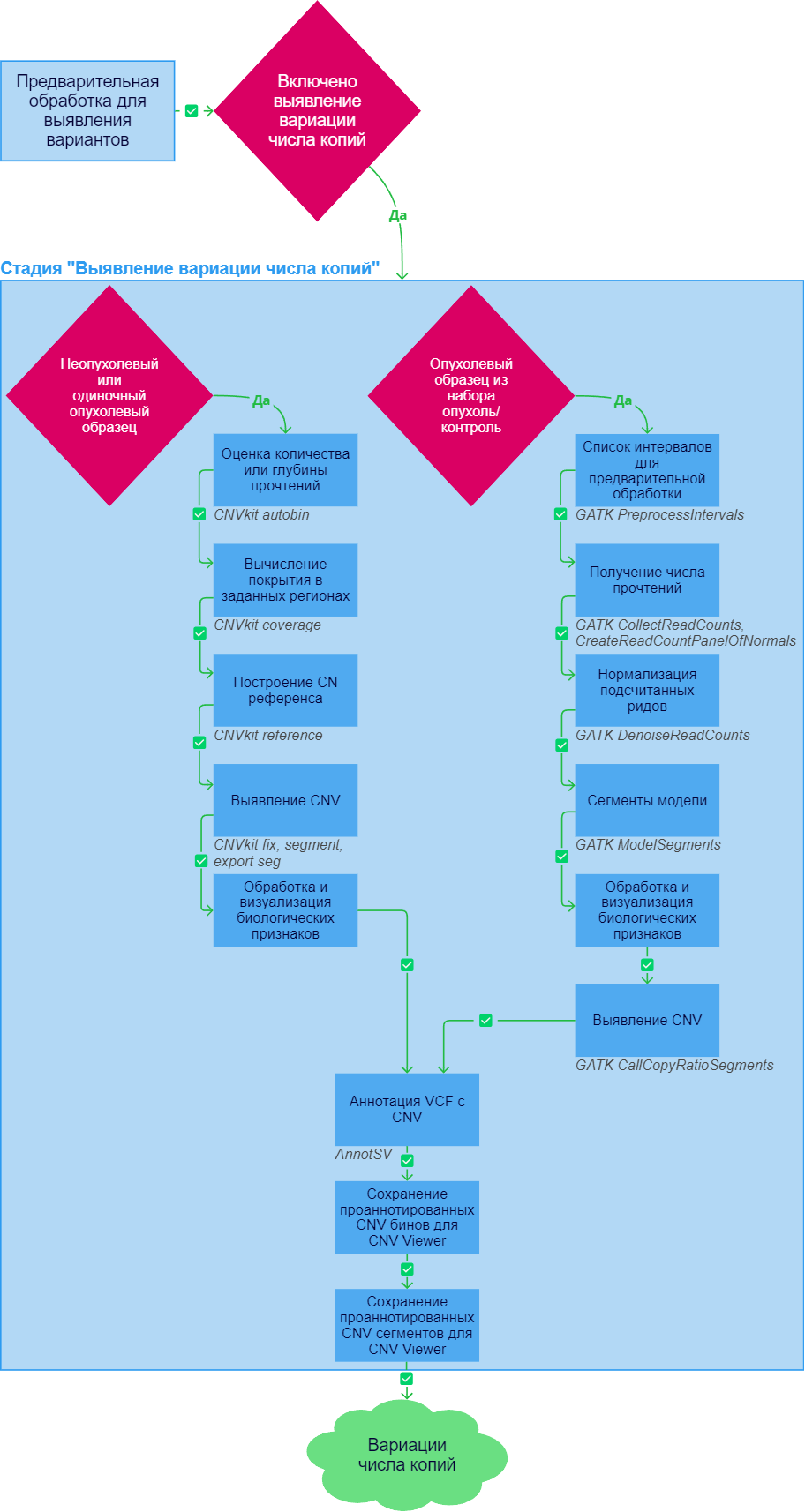
Неопухолевый или одиночный опухолевый образец#
В случае анализа образца секвенирования только опухолевой ткани без контроля или пренатальной диагностики (анализ образца секвенирования неопухолевой ткани) для выявления вариации числа копий используются инструменты CNVkit.
1. Оценка количества или глубины прочтений#
На первом этапе CNVkit autobin оценивает количество или глубину прочтений в файле выравнивания, чтобы выявить оптимальные размеры целевых и антицелевых бинов (интервалов оценки покрытия). При этом используются модели генов, чтобы присвоить названия целевым регионам. В зависимости от глубины покрытия и типа секвенирования требуется разная разбивка генома на регионы, внутри которых будет происходить подсчет прочтений:
- В случае определения типа секвенирования образца как "Панель" (секвенирование с помощью таргетной панели) или "WES" (whole-exome sequencing; полноэкзомное секвенирование) инструменту передаются уникально картируемые регионы генома и соответствующие образцу секвенирования интервалы реагента capture kit - таргетной панели, используемой при направленном отборе.
- В случае определения типа секвенирования образца как "WGS" (whole-genome sequencing; полногеномное секвенирование) или "Low-pass WGS" (полногеномное секвенирование с низким покрытием) инструменту передаются доступные для секвенирования геномные регионы. Сначала рассчитывается пристрелочный размер бина в пределах от 10 до 1000000000 оснований. Затем рассчитанный размер используется в качестве ограничения снизу или сверху размера, значение которого регулирется соответствующей настройкой. Именно этот размер используется при создании итоговых файлов.
В результате autobin формирует целевые и антицелевые BED файлы и выводит таблицу подсчитанных средних глубин покрытия и рекомендуемых размеров бинов.
Если для анализа образца выбрана панель референсных образцов для WGS или панель референсных образцов для направленного отбора, то вместо выявления оптимальных размеров бинов будет использован размер, соответствующий выбранной панели (для некоторых панелей его можно настраивать).
Полученные файлы с целевыми и антицелевыми бинами можно скачать в разделе "Файлы с результатами" в деталях задачи "Оценка количества или глубины прочтений" ("Скачать target BED" и "Скачать antitarget BED", соответственно).
На заметку
Если в ходе анализа было выявлено, что образец секвенирования имеет слишком низкое покрытие реагентом capture kit (более 10% регионов для набора реагентов имеют нулевое покрытие), то дальнейший анализ образца с целью выявления вариации числа копий не выполняется. Такое может произойти, если набор реагентов был выбран неправильно или если образец имеет низкое качество.
2. Вычисление покрытия в заданных регионах#
На данном этапе происходит точный подсчет количества прочтений в каждом из бинов, которые были определены на предыдущем этапе, с помощью CNVkit coverage. Покрытие рассчитывается через среднюю глубину покрытия в бине. Полученные файлы с информацией о покрытии в целевых и антицелевых бинах можно скачать в разделе "Файлы с результатами" в деталях задачи "Вычисление покрытия в заданных регионах" ("Скачать target coverage CNN" и "Скачать antitarget coverage CNN", соответственно).
3. Построение CN референса#
Для определения вариации числа копий по глубине прочтений необходимо оценить отклонение глубины от нормы. Нормальное покрытие в регионах рассчитывается с учетом GC-состава и доли маскировки повторов в регионах. Это осуществляется с помощью CNVkit reference. В рассчёте участвуют как целевые, так и антицелевые бины.
Если для анализа образца выбрана панель референсных образцов для WGS или панель референсных образцов для направленного отбора, то вместо построения референса числа копий будет использована эта панель.
Полученный файл со значениями ожидаемой глубины покрытия и надежности этой оценки можно скачать в разделе "Файлы с результатами" в деталях задачи "Построение CN референса" ("Скачать CNN").
4. Выявление CNV#
Это ключевой этап, на котором рассчитывается количество копий того или иного участка генома на основе количества прочтений, картированных на геном. Для этого используются инструменты CNVkit:
- CNVkit fix объединяет таблицы покрытия целевых и антицелевых бинов и корректирует погрешность в покрытии по регионам и GC-составе согласно референсу. Получившуюся таблицу соотношения числа копий можно скачать в разделе "Файлы с результатами" в деталях задачи "Выявление CNV" ("Скачать CNR").
- CNVkit segment выводит отдельные сегменты числа копий из таблицы покрытия. Если тип секвенирования образца - таргетная панель, либо если анализируется одиночный образец опухолевой ткани, то в качестве алгоритма сегментации используется круговая бинарная сегментация (circular binary segmentation, CBS). Если тип секвенирования был определён как WGS, то в качестве алгоритма сегментации используется экспериментальный метод - скрытая марковская модель с фиксированной амплитудой для состояний делеции, нейтральности и дупликации, соответствующих абсолютному числу копий 1, 2 и 3. Получившийся файл с сегментами можно скачать в том же разделе ("Скачать CNS").
- CNVkit export seg конвертирует файлы сегментации в стандартный формат SEG для загрузки во встроенный модуль визуализации вариаций на геноме Integrative Genomics Viewer (IGV). Выявленные сегменты в формате SEG можно скачать в том же разделе ("Скачать Called segments SEG").
5. Обработка и визуализация биологических признаков#
На данном этапе происходит обработка вариаций числа копий в зависимости от порогового значения log2FC - логарифмического соотношения выявленного числа копий к нормальному числу копий. Пороговое значение по умолчанию (-0,7) можно поменять в параметрах. На его примере посмотрим, как будут рассчитываться пороги для вызова делеций и дупликаций на аутосомах и половых хромосомах:
- Вариации числа копий на аутосомах с log2FC ≤ -0.7 принимаются за делеции (то есть ниже этого порога вызывается одна аутосома вместо двух в норме).
- Вариации числа копий на X- и Y-хромосомах с log2FC ≤ -2.11 принимаются за делеции.
- Вариации числа копий на аутосомах с log2FC ≥ 0.46 принимаются за дупликации. Порог для дупликации на аутосоме рассчитывается по следующей формуле: log2FC = log2((2+D)/2), где D - "количество" копийности, которое "теряется" при log2FC=-0.7, а 2 - количество сестринских хроматид в аутосоме.
- Вариации числа копий на X- и Y-хромосомах с log2FC ≥ 0.82 принимаются за дупликации.
- Вариации, не удовлетворяющие порогам, т.е. вариации на аутосомах с -0.7 < log2FC < 0.46 и вариации на половых хромосомах с -2.11 < log2FC < 0.82, не проходят фильтрацию.
Нажмите, чтобы посмотреть, как производится расчёт порогов log2FC для делеций и дупликаций на аутосомах и половых хромосомах
Дельта числа копий: D = 2 – C = 2 – 2 × 2T21.
Тогда соответствующий порог вызова трёх копий вместо двух на аутосомах вычисляется так: T23 = log2((2+D)/2) = log2(2-2T21).
Порог для вызова делеций на аутосомах: lo = T21.
Порог для вызова дупликаций на аутосомах: hi = T23.
Для вычисления порога для вызова трёх копий вместо двух на Х-хромосоме нормализуем генотип XY, поделив его на число копий, равное 1: T23X= log2((2+D)/1) = log2(2+2-2×2T21) = 1 + log2(2-2T21) = 1 + T23.
Вычисление порога для вызова дупликаций на X-хромосоме:
- если нет Y-хромосомы: Xhi = 1 + T23;
- если есть Y-хромосома: Xhi = log2(3-2×2T21).
Порог для вызова одной копии вместо двух на Х-хромосоме: T21X = log2((2-D)/1) = log2(2-2+2×2T21) = 1 + T21.
Порог для вызова ни одной копии вместо одной на X-хромосоме: T10X = log2((1-D)/1) = log2(1-2+2×2T21) = log2(-1+2×2T21).
Вычисление порога для вызова делеций на X-хромосоме:
- если нет Y-хромосомы: Xlo = 1 + T21;
- если есть Y-хромосома: Xlo = log2(-1+2×2T21).
Порог для вызова одной копии вместо ни одной на Y-хромосоме в случае генотипа XX: T01 = log2((0+D)/1) = log2(2-2×2T21) = log2(2×(1-2T21)) = 1 + log2(1-2T21).
Вычисление порога для вызова дупликаций Yhi и делеций Ylo на Y-хромосоме:
- если нет Y-хромосомы: Yhi = 1 + log2(1-2T21)); Ylo = -∞;
- если есть Y-хромосома: Yhi = Xhi; Ylo = Xlo.
Если для образца неопухолевой ткани активирован поиск только герминальных анеуплоидий, то в итоговых файлах будут представлены только анеуплоидии (вариации числа копий хромосом).
В результате обработки вариаций создаются следующие файлы, которые можно скачать в разделе "Файлы с результатами" в деталях задачи "Обработка и визуализация биологических признаков":
- Файл покрытия CNV в формате TSV - неаннотированный файл, удобный для рассмотрения изменения копийности на крупных участках (плечи хромосом, хромосомы). Ссылка для скачивания файла: "Скачать CNV coverage TSV". Этот файл можно также открыть в таблицах Google.
- Файл с бинами числа копий в формате BED ("Скачать CNV bins BED").
- Файл с сегментами числа копий в формате BED ("Скачать CNV segments BED").
- Файл с порогами log2FC - логарифмического соотношения выявленного числа копий к нормальному числу копий, на основе которых происходит фильтрация вариаций числа копий. Отдельно приведены значения log2FC, при которых вариации на аутосомах, X-хромосоме или Y-хромосоме принимаются за делеции или дупликации. Ссылка для скачивания файла: "Скачать CNV thresholds JSON".
Кроме того, формируются файлы с полногеномной визуализацией и кариограммой, удобные для оценки крупных изменений копийности. Их можно открыть на странице результатов "Главное" или скачать в разделе "Файлы с результатами" в деталях задачи "Обработка и визуализация биологических признаков":
- График по типу кариограммы с обозначенными CNV уровня хромосом в формате JPG ("Скачать Karyogram-like graph JPG").
- Полногеномный график сегментов CNV в формате JPG ("Скачать JPG").
- Файл в формате PDF с графиками сегментов CNV по всем хромосомам ("Скачать PDF").
- Файлы с графиками сегментов CNV отдельно для каждой хромосомы ("Скачать [хромосома] JPG").
6. Аннотация VCF с CNV#
Для полной и эффективной интерпретации влияния CNV на фенотип каждая найденная вариация аннотируется. Аннотирование и ранжирование выявленных вариаций числа копий осуществляется программой AnnotSV. Она собирает функционально, регуляторно и клинически значимую информацию и направлена на предоставление аннотаций, полезных для интерпретации потенциальной патогенности вариаций и фильтрации потенциальных ложноположительных результатов. Аннотация производится отдельно для сегментов и бинов.
Получившиеся файлы с проаннотированными вариациями числа копий можно скачать в разделе "Файлы с результатами" в деталях задачи "Аннотация VCF с CNV":
- Файл со всеми сегментами числа копий в формате TSV - "Скачать Segments all TSV". Его также можно открыть в таблицах Google.
- Файл со всеми сегментами числа копий в формате CSV - "Скачать Segments all CSV".
- Файл с отфильтрованными сегментами числа копий в формате TSV - "Скачать Segments filtered TSV". Его также можно открыть в таблицах Google.
- Файл с отфильтрованными сегментами числа копий в формате CSV - "Скачать Segments filtered CSV".
- Файл со всеми бинами числа копий в формате TSV - "Скачать Bins all TSV". Его также можно открыть в таблицах Google.
- Файл со всеми бинами числа копий в формате CSV - "Скачать Bins all CSV".
- Файл с отфильтрованными бинами числа копий в формате TSV - "Скачать Bins filtered TSV". Его также можно открыть в таблицах Google.
- Файл с отфильтрованными бинами числа копий в формате CSV - "Скачать Bins filtered CSV".
С форматом и полями аннотации можно ознакомиться в документации инструмента AnnotSV.
7. Сохранение проаннотированных CNV бинов для CNV Viewer#
Сохранение результатов выявления вариации числа копий в бинах для показа во встроенном модуле для просмотра и анализа вариаций CNV Viewer.
8. Сохранение проаннотированных CNV сегментов для CNV Viewer#
Сохранение результатов выявления вариации числа копий в сегментах для показа во встроенном модуле для просмотра и анализа вариаций CNV Viewer.
После стадии "Выявление вариации числа копий" анализ может продолжиться генерацией отчётов.
Опухолевый образец из набора опухоль/контроль#
В случае анализа образца секвенирования опухолевой ткани на фоне здоровой для выявления вариации числа копий применяется пайплайн GATK.
1. Список интервалов для предварительной обработки#
GATK PreprocessIntervals подготавливает бины (интервалы оценки покрытия) для сбора информации о покрытии. Подготовка бинов различается в зависимости от типа секвенирования образца:
- В случае определения типа секвенирования образца как "Панель" (секвенирование с помощью таргетной панели) или "WES" (whole-exome sequencing; полноэкзомное секвенирование) входные интервалы (capture kit) сначала проверяются на наличие перекрывающихся интервалов, которые объединяются. Затем полученные интервалы дополняются и разделяются на бины. Бины, содержащие только N (неизвестные нуклеотиды), отфильтровываются. Биннинг (группировка интервалов в меньшее количество бинов) не производится. Примыкающие интервалы (т.е. интервалы, которые непосредственно находятся рядом, но фактически не перекрываются) не объединяются, а обрабатываются как отдельные интервалы.
- В случае определения типа секвенирования образца как "WGS" (whole-genome sequencing; полногеномное секвенирование) или "Low-pass WGS" (полногеномное секвенирование с низким покрытием) каждый контиг принимается за один интервал и соответствующим образом группируется. Примыкающие интервалы объединяются. Это создает бины, подходящие для анализа последовательности всего генома. Длина генерируемых последовательных бинов регулируется соответствующей настройкой. Бины, содержащие только N (неизвестные нуклеотиды), отфильтровываются.
Полученный предварительно обработанный файл Picard со списком интервалов можно скачать в разделе "Файлы с результатами" в деталях задачи "Список интервалов для предварительной обработки" ("Скачать INTERVAL_LIST").
2. Получение числа прочтений#
GATK CollectReadCounts собирает число прочтений в указанных интервалах. Число для каждого интервала вычисляется путём подсчёта количества стартов прочтений, находящихся в интервале. Примыкающие интервалы не объединяются, а обрабатываются как отдельные интервалы. Команда выполняется для файлов выравниваний опухолевого и соответствующего контрольного образцов. Получившиеся файлы с числом прочтений в формате HDF5 (Hierarchical Data Format, Version 5) можно скачать в разделе "Файлы с результатами" в деталях задачи "Получение числа прочтений" (результат для контрольного образца - "Скачать Normal file read counts HDF5", результат для опухолевого образца - "Скачать Tumor file read counts HDF5").
GATK CreateReadCountPanelOfNormals создаёт нормальную панель (Panel of Normal; PON) для удаления шума в подсчёте прочтений, учитывая число прочтений для образцов в панели. Проводится на основе результата подсчёта числа прочтений в контрольном образце. Геномные интервалы с медианой фракционного покрытия (по всем образцам), меньшей либо равной пятому процентилю, отфильтровываются. Файл с созданной панелью можно скачать в разделе "Файлы с результатами" в деталях задачи "Получение числа прочтений" ("Скачать Generated panel of normal HDF5").
3. Нормализация подсчитанных ридов#
GATK DenoiseReadCounts очищает от шума число прочтений для получения соотношений копий, очищенных от шума. Полученная на предыдущем этапе нормальная панель используется для стандартизации и уменьшения шума числа прочтений, собранных в опухолевом образце. Стандартизация включает: (a) преобразование входного числа прочтений в фракционное покрытие; (b) фильтрацию интервалов до тех, которые содержатся в панели; (c) деление числа прочтений на медианы интервалов, входящих в панель; (d) деление на медиану образца; (e) преобразование в log2 соотношения копий. Затем результат очищается от шума путем вычитания проекции на указанное количество главных компонент из панели. На выходе получаются следующие файлы, которые можно скачать в разделе "Файлы с результатами" в деталях задачи "Нормализация подсчитанных ридов":
- Файл с соотношениями копий, очищенными от шума, в формате TSV - "Скачать Denoised copy ratios TSV". Его можно также открыть в таблицах Google.
- Файл со стандартизированными соотношениями копий в формате TSV - "Скачать Standardized copy ratios TSV". Его можно также открыть в таблицах Google.
4. Сегменты модели#
GATK ModelSegments моделирует сегментированные соотношения копий на основе файла с соотношениями копий, очищенными от шума. Сегментация происходит с помощью метода, основанного на алгоритме сегментации ядра. Затем используется метод Монте-Карло с марковскими цепями, чтобы определить апостериорные значения сегментированных моделей для log2 соотношения копий. Наконец, выполняется сглаживание сегментированных апостериорных значений путём слияния смежных сегментов, чьи апостериорные доверительные интервалы в достаточной мере перекрываются (количество 10%-ной ширины доверительного интервала с равными концами должно равняться 2). Сглаживание может повторяться несколько раз до конвергенции, после чего вновь используется метод Монте-Карло с марковскими цепями.
Файл, содержащий сегменты соотношения копий, можно скачать в разделе "Файлы с результатами" в деталях задачи "Сегменты модели" ("Скачать Copy ration segments SEG"). Файл со смоделированными сегментами после сглаживания сегментации можно скачать в том же разделе ("Скачать Modeled segments SEG").
5. Обработка и визуализация биологических признаков#
На данном этапе происходит обработка вариаций числа копий в зависимости от порогового значения log2FC - логарифмического соотношения выявленного числа копий к нормальному числу копий. Пороговое значение по умолчанию (-0,7) можно поменять в параметрах. На его примере посмотрим, как будут рассчитываться пороги для вызова делеций и дупликаций на аутосомах и половых хромосомах:
- Вариации числа копий на аутосомах с log2FC ≤ -0.7 принимаются за делеции (то есть ниже этого порога вызывается одна аутосома вместо двух в норме).
- Вариации числа копий на X- и Y-хромосомах с log2FC ≤ -2.11 принимаются за делеции.
- Вариации числа копий на аутосомах с log2FC ≥ 0.46 принимаются за дупликации. Порог для дупликации на аутосоме рассчитывается по следующей формуле: log2FC = log2((2+D)/2), где D - "количество" копийности, которое "теряется" при log2FC=-0.7, а 2 - количество сестринских хроматид в аутосоме.
- Вариации числа копий на X- и Y-хромосомах с log2FC ≥ 0.82 принимаются за дупликации.
- Вариации, не удовлетворяющие порогам, т.е. вариации на аутосомах с -0.7 < log2FC < 0.46 и вариации на половых хромосомах с -2.11 < log2FC < 0.82, не проходят фильтрацию.
Нажмите, чтобы посмотреть, как производится расчёт порогов log2FC для делеций и дупликаций на аутосомах и половых хромосомах
Дельта числа копий: D = 2 – C = 2 – 2 × 2T21.
Тогда соответствующий порог вызова трёх копий вместо двух на аутосомах вычисляется так: T23 = log2((2+D)/2) = log2(2-2T21).
Порог для вызова делеций на аутосомах: lo = T21.
Порог для вызова дупликаций на аутосомах: hi = T23.
Для вычисления порога для вызова трёх копий вместо двух на Х-хромосоме нормализуем генотип XY, поделив его на число копий, равное 1: T23X= log2((2+D)/1) = log2(2+2-2×2T21) = 1 + log2(2-2T21) = 1 + T23.
Вычисление порога для вызова дупликаций на X-хромосоме:
- если нет Y-хромосомы: Xhi = 1 + T23;
- если есть Y-хромосома: Xhi = log2(3-2×2T21).
Порог для вызова одной копии вместо двух на Х-хромосоме: T21X = log2((2-D)/1) = log2(2-2+2×2T21) = 1 + T21.
Порог для вызова ни одной копии вместо одной на X-хромосоме: T10X = log2((1-D)/1) = log2(1-2+2×2T21) = log2(-1+2×2T21).
Вычисление порога для вызова делеций на X-хромосоме:
- если нет Y-хромосомы: Xlo = 1 + T21;
- если есть Y-хромосома: Xlo = log2(-1+2×2T21).
Порог для вызова одной копии вместо ни одной на Y-хромосоме в случае генотипа XX: T01 = log2((0+D)/1) = log2(2-2×2T21) = log2(2×(1-2T21)) = 1 + log2(1-2T21).
Вычисление порога для вызова дупликаций Yhi и делеций Ylo на Y-хромосоме:
- если нет Y-хромосомы: Yhi = 1 + log2(1-2T21)); Ylo = -∞;
- если есть Y-хромосома: Yhi = Xhi; Ylo = Xlo.
В результате обработки вариаций создаются следующие файлы, которые можно скачать в разделе "Файлы с результатами" в деталях задачи "Обработка и визуализация биологических признаков":
- Файл покрытия CNV в формате TSV - неаннотированный файл, удобный для рассмотрения изменения копийности на крупных участках (плечи хромосом, хромосомы). Ссылка для скачивания файла: "Скачать CNV coverage TSV". Этот файл можно также открыть в таблицах Google.
- Файл с бинами числа копий в формате BED ("Скачать CNV bins BED").
- Файл с сегментами числа копий в формате BED ("Скачать CNV segments BED").
- Файл с порогами log2FC - логарифмического соотношения выявленного числа копий к нормальному числу копий, на основе которых происходит фильтрация вариаций числа копий. Отдельно приведены значения log2FC, при которых вариации на аутосомах, X-хромосоме или Y-хромосоме принимаются за делеции или дупликации. Ссылка для скачивания файла: "Скачать CNV thresholds JSON".
Кроме того, формируются файлы с полногеномной визуализацией и кариограммой, удобные для оценки крупных изменений копийности. Их можно открыть на странице результатов "Главное" или скачать в разделе "Файлы с результатами" в деталях задачи "Обработка и визуализация биологических признаков":
- График по типу кариограммы с обозначенными CNV уровня хромосом в формате JPG ("Скачать Karyogram-like graph JPG").
- Полногеномный график сегментов CNV в формате JPG ("Скачать JPG").
- Файл в формате PDF с графиками сегментов CNV по всем хромосомам ("Скачать PDF").
- Файлы с графиками сегментов CNV отдельно для каждой хромосомы ("Скачать [хромосома] JPG").
6. Выявление CNV#
Это ключевой этап, на котором рассчитывается количество копий того или иного участка генома на основе количества прочтений, картированных на геном. В случае анализа образца секвенирования опухолевой ткани на фоне здоровой применяется пайплайн GATK.
Сегменты соотношения копий распознаются как амплифицированные, делетированные или нейтральные по числу копий с помощью GATK CallCopyRatioSegments. Это относительно наивный коллер, который берёт сегменты, смоделированные на этапе "Сегменты модели", и выполняет простой статистический тест на сегментированных log2 соотношениях копий для распознавания амплификации и делеции, учитывая заданный диапазон для определения нейтральных по числу копий сегментов.
На выходе получаются следующие файлы, которые можно скачать в разделе "Файлы с результатами" в деталях задачи "Выявление CNV":
- Файл с распознанными сегментами соотношения копий в формате TSV - "Скачать Called segments SEG".
- Файл CBS (circular binary segmentation) с информацией о сегментах - "Скачать Statistics SEG". Он совместим со встроенным модулем визуализации вариаций на геноме Integrative Genomics Viewer (IGV). С описанием формата файла и его колонок можно ознакомиться здесь.
7. Аннотация VCF с CNV#
Для полной и эффективной интерпретации влияния CNV на фенотип каждая найденная вариация аннотируется. Аннотирование и ранжирование выявленных вариаций числа копий осуществляется программой AnnotSV. Она собирает функционально, регуляторно и клинически значимую информацию и направлена на предоставление аннотаций, полезных для интерпретации потенциальной патогенности вариаций и фильтрации потенциальных ложноположительных результатов. Аннотация производится отдельно для сегментов и бинов.
Получившиеся файлы с проаннотированными вариациями числа копий можно скачать в разделе "Файлы с результатами" в деталях задачи "Аннотация VCF с CNV":
- Файл со всеми сегментами числа копий в формате TSV - "Скачать Segments all TSV". Его также можно открыть в таблицах Google.
- Файл со всеми сегментами числа копий в формате CSV - "Скачать Segments all CSV".
- Файл с отфильтрованными сегментами числа копий в формате TSV - "Скачать Segments filtered TSV". Его также можно открыть в таблицах Google.
- Файл с отфильтрованными сегментами числа копий в формате CSV - "Скачать Segments filtered CSV".
- Файл со всеми бинами числа копий в формате TSV - "Скачать Bins all TSV". Его также можно открыть в таблицах Google.
- Файл со всеми бинами числа копий в формате CSV - "Скачать Bins all CSV".
- Файл с отфильтрованными бинами числа копий в формате TSV - "Скачать Bins filtered TSV". Его также можно открыть в таблицах Google.
- Файл с отфильтрованными бинами числа копий в формате CSV - "Скачать Bins filtered CSV".
С форматом и полями аннотации можно ознакомиться в документации инструмента AnnotSV.
8. Сохранение проаннотированных CNV бинов для CNV Viewer#
Сохранение результатов выявления вариации числа копий в бинах для показа во встроенном модуле для просмотра и анализа вариаций CNV Viewer.
9. Сохранение проаннотированных CNV сегментов для CNV Viewer#
Сохранение результатов выявления вариации числа копий в сегментах для показа во встроенном модуле для просмотра и анализа вариаций CNV Viewer.
После стадии "Выявление вариации числа копий" анализ может продолжиться генерацией отчётов.