Calculate coverage
At this stage, the coverage of genes and target regions (capture kit intervals) by sample read alignments is calculated. These metrics are important for assessing the quality of mapping in general and in the regions of interest (genes, transcripts). The coverage calculation results will be located on the "Coverage Analysis" tab on the sample page. The coverage calculation is launched after the "Variant discovery preprocessing" stage has been successfully completed.
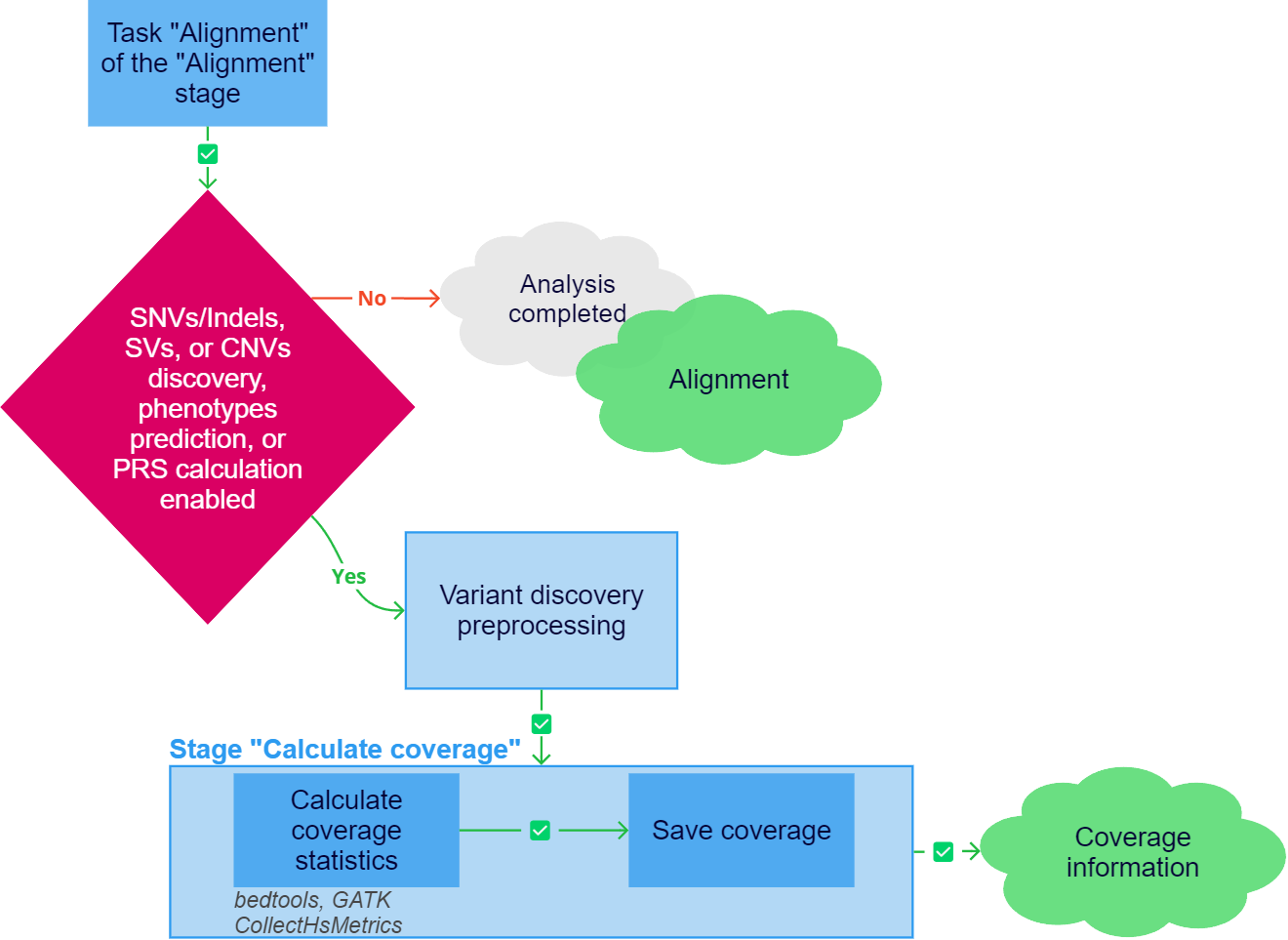
The "Calculate coverage" stage of sample analysis includes the following tasks:
Calculate coverage statistics. The workflow of the task depends on the sample sequencing type:
1.1. If the sequencing type is defined as Targeted selection and a capture kit is defined for the sample, the task includes the following steps:
- bedtools coverage computes the depth and breadth of coverage of sample read alignments on capture kit intervals and reports a histogram of coverage for each capture kit interval as well as a summary histogram for all intervals.
- bedtools intersect screens for overlaps between exons from Ensembl and capture kit intervals.
- bedtools coverage computes the depth and breadth of coverage of sample read alignments on exons from Ensembl and reports a histogram of coverage for each exon as well as a summary histogram for all exons.
The file with gene coverage on mapped reads can be downloaded in the "Result files" section in the "Calculate coverage statistics" task details ("Download Gene coverage TSV"). You can also open it in Google Sheets.
GATK CollectHsMetrics collects hybrid-selection (HS) metrics for the sample alignment file within capture kit intervals.
The file with the calculated metrics can be downloaded in the "Result files" section in the "Calculate coverage statistics" task details ("Download HS Metrics TSV"). You can also open it in Google Sheets.
The file with information about the coverage of target regions (capture kit intervals) can be downloaded in the same section ("Download Per target coverage TSV"). You can also open it in Google Sheets.1.2. If the sequencing type is defined as Targeted selection, but a capture kit is not defined for the sample, the task includes the following step:
bedtools coverage computes the depth and breadth of coverage of sample read alignments on exons from Ensembl and reports a histogram of coverage for each exon as well as a summary histogram for all exons. The file with gene coverage on mapped reads can be downloaded in the "Result files" section in the "Calculate coverage statistics" task details ("Download Gene coverage TSV"). You can also open it in Google Sheets.
1.3. If the sequencing type is defined as WGS (whole-genome sequencing), the task includes the following steps:
bedtools coverage computes the depth and breadth of coverage of sample read alignments on exons from Ensembl and reports a histogram of coverage for each exon as well as a summary histogram for all exons.
bedtools genomecov computes histograms of aligned sample sequences coverage for the reference genome.
The file with gene coverage on mapped reads can be downloaded in the "Result files" section in the "Calculate coverage statistics" task details ("Download Gene coverage TSV"). You can also open it in Google Sheets.
- Save coverage task is saving of gene and transcript coverage.