Upload, identify and verify
If a single read sequencing sample is uploaded, only one file will be processed and the initial workflow stage will be called "Upload, identify and verify". If a paired read sequencing sample is uploaded, then two paired files will be analyzed and there will be two initial workflow stages: "Upload, identify and verify primary" and "Upload, identify and verify mate".
During the initial workflow stage, the FASTQ or BAM format sample file(s) are uploaded, their format is identified, and they are verified. If any of the following tasks fail, the sample analysis is stopped.
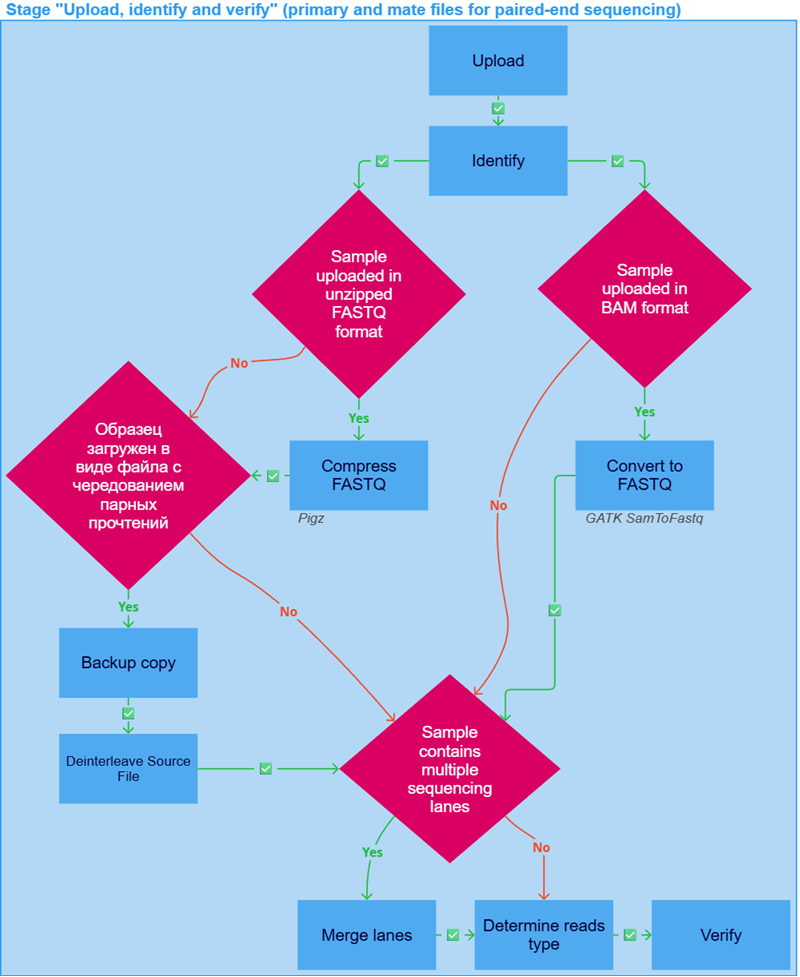
The "Upload, identify and verify" stage of sample analysis may include the following tasks:
- Upload. If you are uploading the sample file(s) from your computer and not via a link, the uploading process may be interrupted. To restore it, use the resume upload form.
- Identify: determination of data format and sequencer. A sample uploaded in BAM format is checked to appear to be intact by samtools quickcheck.
- Compress FASTQ to GZIP archive if the sample was uploaded as a FASTQ file, not packed into an archive or packed into an archive other than GZIP (e.g. ZIP, BZIP2, 7-ZIP, XZ, WIM, RAR). Compression is performed using Pigz.
- Convert to FASTQ if the sample was uploaded in BAM format. It is performed using GATK SamToFastq. The resulting FASTQ files are compressed into a GZIP archive using Pigz. The original BAM file can be downloaded in the "Result files" section in the "Convert to FASTQ" task details ("Download Original BAM").
- The sample can be uploaded as an interleaved file. In such a file, the primary read is always written right before the corresponding mate read for each pair of reads. Such a sample undergoes additional pre-analysis tasks, such as Backup cop and Deinterleave Source File, which are necessary to accurately split the reads from the file into primary and mate reads and write them to two separate files. All pre-split tasks except "Identify" will be recorded in the details of the "Upload, identify and verify primary" stage, and the "Upload, identify and verify mate" stage will include only the "Identify", "Determine reads type" and "Verify" tasks.
- Merge lanes is the process of combining multiple FASTQ files into a single sample, in cases where the sample was either automatically recognized or manually specified during upload as a sequencing sample consisting of multiple lanes from a single flow cell.
- Determine reads type is an experimental feature that does not affect the downstream analysis.
This task uses a built-in classifier based on an LSTM (long short-term memory) model to predict the type of nucleic
acid (DNA, RNA, or UNKNOWN) whose sequence was obtained during sample preparation. The predicted result is displayed
on the sample page, where it can be manually corrected if needed.
The "Determine reads type" task is not executed if a settings preset with a predefined reads type (DNA or RNA) was selected during sample upload. In this case, the task is included in the pipeline but not run, and the specified type is set without additional validation. - Verify file integrity; check that there are four lines per read; check that the lengths of sequence and sequence qualities are the same.
The uploaded compressed FASTQ sample file(s) can be downloaded at the top of the "Workflow details" tab. In the case of an interleaved paired read sample, you can download there the primary and mate read files after splitting.
After the "Upload, identify and verify" stage has successfully completed, the analysis continues with quality control.