Variant discovery preprocessing
After the "Alignment" task of the stage of the same name has been successfully completed, it is recommended to further preprocess the alignment results for the most accurate variants discovery. This increases sensitivity to rare variants, contributes to a more accurate copy number count, which depends on the number of reads, and also speeds up sample processing. Variant discovery preprocessing is launched if at least one of the following stages is included in the sample analysis workflow: somatic SNVs/Indels discovery, germline SNVs/Indels discovery, structural variations discovery, copy number variations discovery, oligogenic risk scores calculation, polygenic risk scores calculation, pharmacogenetic calculation, or ancestry analysis. If any of the tasks listed below fails, the sample analysis is stopped.
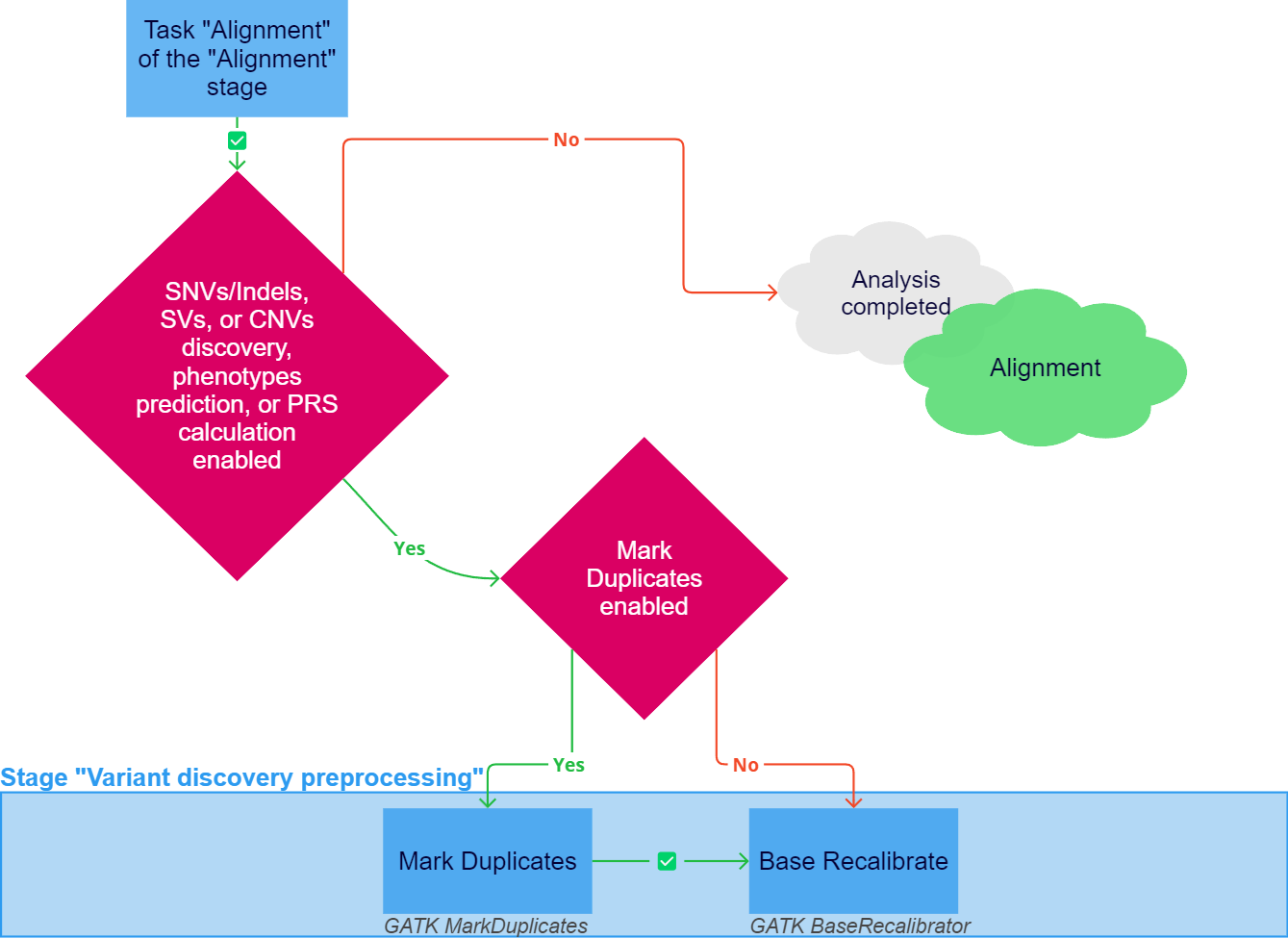
The "Variant discovery preprocessing" stage of sample analysis may include the following tasks:
Mark Duplicates if the corresponding parameter is enabled for the sample. During sequencing, the amount of original sequences of fragmented DNA increases in the polymerase chain reaction (PCR). For further analysis, it is very important to take into account duplicate reads of the same fragment. If you want to keep all reads for further analysis (for example, if you are working with amplicons), you can disable the marking of duplicates.
Marking duplicates is performed using the GATK MarkDuplicates tool, which locates and tags duplicate reads in the alignment file, where duplicate reads are defined as originating from a single fragment of DNA. Further analysis is performed only on the original reads. The resulting file in BAM format is sorted by coordinates and an index file is created for it. In addition, a file with metrics is created, which indicates the number of fragments or read pairs that were marked as duplicates. This file can be downloaded in the "Result files" section in the "Mark Duplicates" task details ("Download duplication metrics TXT").Base Recalibrate. The sequencers assign a quality score to each nucleotide, which is the log likelihood of a nucleotide occurrence at a given position compared to other nucleotides. However, many sequencers tend to underestimate sequencing quality and make other errors. Base recalibration corrects for the systematic bias that affects the sequencer's assignment of quality score to nucleotides, which reduces error and improves the quality of further variant discovery.
Base Quality Score Recalibration (BQSR) is performed in three steps:
- GATK BaseRecalibrator calculates an empirical probability of error and finds patterns in how error varies with nucleotide calling patterns across all nucleotides. These observations are written to a recalibration table based on such covariates as read group, reported quality score, machine cycle, and nucleotide context. BaseRecalibrator does a by-locus traversal operating only within certain intervals at known polymorphic sites used to exclude regions around known polymorphisms from analysis. All reference mismatches are considered errors and indicative of poor base quality. An empirical probability of error (p) is then calculated given the particular covariates seen at this site, where p = num mismatches / num observations. The output file is a table containing the values of the individual covariates, number of observations for this combination of covariates, number of reference mismatches, and the raw empirical quality score calculated by phred-scaling the mismatch rate. The recalibration table can be downloaded in the "Result files" section in the "Base Recalibrate" task details ("Download recalibration table TXT").
- GATK ApplyBQSR applies base quality score recalibration within certain intervals, i.e. recalibrates the base qualities of the input reads based on the recalibration table produced in the first step, and outputs the recalibrated BAM files.
- GATK MergeSamFiles merges BAM files recalibrated within certain intervals into a single file. The resulting BAM file is sorted by coordinates and an index file is created for it.
The final sample alignment file after preprocessing can be downloaded in the "Result files" section in the "Base Recalibrate" task details ("Download BAM"). You can also open this file in the IGV browser by clicking on the "Open in IGV Browser" link. The index file for the final BAM file can be downloaded there as well ("Download BAI").
info
If you want to add the alignment track obtained after preprocessing your sample in Genomenal to your desktop IGV, you can do so via a link. To do this, follow these steps:
- Right-click the alignment file link "Download BAM" and select the "Copy link address" option.
- Upload the track via URL to your desktop IGV, as described here.
- Right-click the alignment index file link "Download BAI" and select the "Copy link address" option.
- Add the index via URL to the corresponding field in IGV.
- Click "OK". Done! The preprocessed sample alignment track is added to IGV.
After the "Variant discovery preprocessing" stage is successfully completed, the analysis continues with coverage calculation and the following stages, if included in the workflow: "Somatic SNVs/Indels discovery" (for a tumor sample), "Germline SNVs/Indels discovery" (for a single tumor sample or a non-tumor sample), "Structural variations discovery", "Copy number variations discovery" and/or "Genomic predictions" (for a non-tumor sample).