Панель детальной информации о варианте
Нажмите на строку варианта, чтобы увидите панель с детальной информацией о варианте под таблицей:
Размер панели можно менять, наведя курсор на верхнюю границу панели и потянув вниз или вверх.
Также панель можно скрыть и развернуть, нажав на соответствующие
стрелки ![]() или
или ![]() .
.
На заметку
На странице "Настройки пользователя" можно отрегулировать, будет ли панель детальной информации о варианте развёрнута или скрыта в SNV Viewer по умолчанию.
Вкладки панели детальной информации#
Панель разделена на 13 вкладок (или 12, если в системе нет ни одной пользовательской аннотации):
- Common
- Gene
- Phenotypes
- ClinVar
- gnomAD 3
- gnomAD 4
- ExAC
- Other frequencies
- Conservation
- Protein function effect
- Protein function effect (additional)
- Other
- Custom annotation sources (если хотя бы одна пользовательская аннотация была добавлена в систему до того, как варианты в образце были проаннотированы)
На панели визуально выделяются только те вкладки, которые включают хотя бы одно не пустое поле аннотации
варианта. Например, вкладка "ClinVar", наполненная
данными, выглядит так:  , а пустая - так:
, а пустая - так:  .
.
По вкладкам панели детальной информации можно перемещаться с помощью горячих клавиш клавиатуры. При этом перемещение будет осуществляться только по непустым вкладкам панели.
Доступные горячие клавиши
| Нажмите эту клавишу | Для этого |
| D или ➡️ | Перемещение на следующую вкладку панели детальной информации, которая имеет хотя бы одно не пустое поле аннотации варианта |
| A или ⬅️ | Перемещение на предыдущую вкладку панели детальной информации, которая имеет хотя бы одно не пустое поле аннотации варианта |
При перемещении между вариантами в таблице выбранная вкладка панели сохраняется.
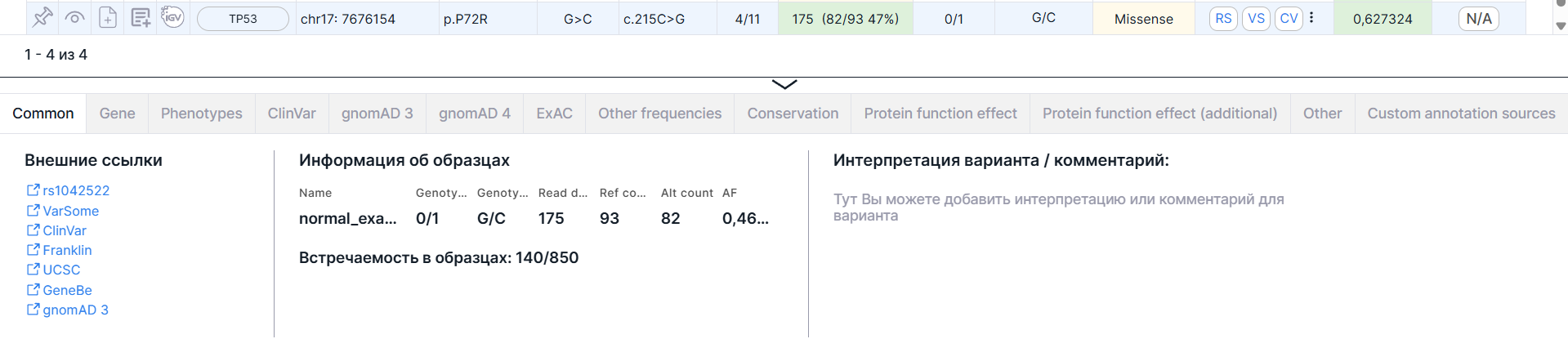
Common#
Вкладка содержит основную информацию о варианте: внешние ссылки, информацию об образцах и интерпретацию варианта. Также может быть приведена информация о нормализованном варианте и фазовой группе.
Внешние ссылки#
Ссылки на страницы с информацией о варианте в базах данных dbSNP (rsId), VarSome, ClinVar, COSMIC (если база была загружена в виде пользовательской аннотации), Franklin, UCSC, gnomAD 3, RUSeq (если база была загружена в виде пользовательской аннотации).
Информация об образцах#
- Таблица с образцами со следующими колонками:
- Name - имя образца, для которого проводился анализ.
- Genotype - генотип, комбинация аллелей для данного образца (0 - референсный аллель, 1 - первый альтернативный аллель, 2 - второй альтернативный аллель и т.д.), разделенных ”/” (для нефазированного генотипа) или “|” (для фазированного генотипа). Количество аллелей говорит о плоидности образца.
- Read depth - глубина секвенирования, общее количество прочтений последовательности, перекрывающих позицию варианта, для данного образца.
- Ref count - количество раз, когда в последовательности считывался референсный аллель для данного образца.
- Alt count - количество раз, когда в последовательности считывался альтернативный аллель для данного образца.
- AF - частота альтернативного аллеля для данного образца.
- Информация о встречаемости варианта в других образцах, загруженных пользователем в систему: количество вариантов/количество образцов. Здесь приведено количество вариантов, которые полностью совпадают с искомым вариантом по типу мутации (соматический или герминальный), хромосоме, стартовой позиции, референсному и альтернативному аллелю и гену, причем учитываются только варианты, которые содержат хотя бы один альтернативный аллель (т.е. имеют генотип 1/1, 1|1, 0/1, 1/0, 0|1, 1|0, 1/., 1|., ./1, .|1, 1, 0/0/1, 0/2, 1/2 и пр.) Эти варианты ищутся среди всех загруженных в систему образцов (включая удаленные), у которых была успешно завершена стадия выявления вариантов (или стадия аннотации вариантов для образцов, загруженных в формате VCF). Для подсчета встречаемости соматического варианта учитываются те образцы, у которых успешно завершена стадия "Выявление соматических SNVs/Indels" (или "Аннотация соматических SNVs/Indels" для образцов в формате VCF), а для подсчета встречаемости герминального варианта - образцы с успешно завершенной стадией "Выявление герминальных SNVs/Indels" (или "Аннотация герминальных SNVs/Indels" для образцов в формате VCF). Нажав на ссылку, вы попадете на вкладку "Встречаемость" страницы детальной информации о варианте.
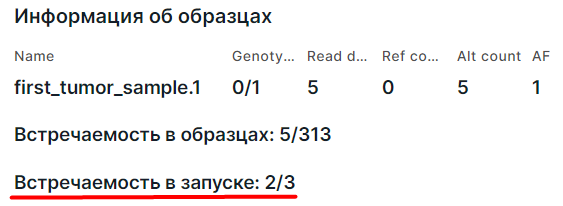
- Информация о встречаемости варианта в образцах запуска. Представлена только на панели вариантов в SNV Viewer запуска и в SNV Viewer образцов запуска. Например, в следующем случае вариант был выявлен в двух из трёх образцов запуска:
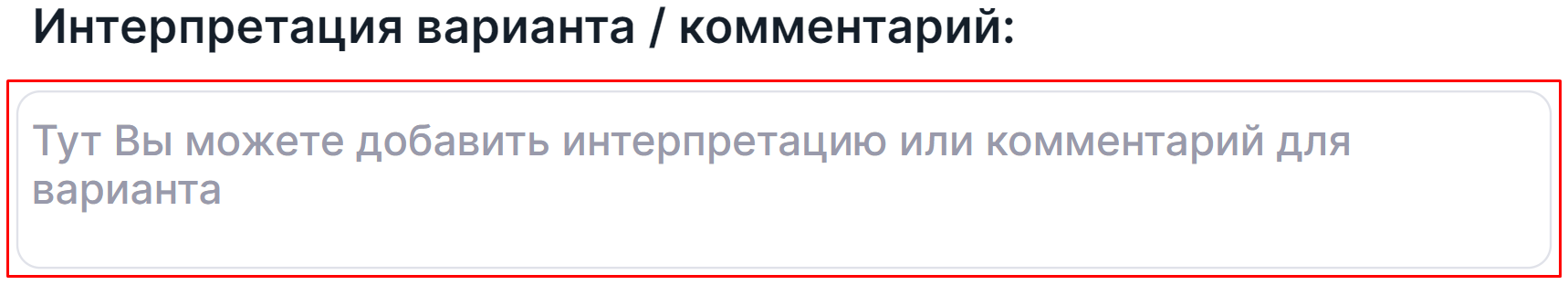
Интерпретация варианта / комментарий#
Интерпретация варианта - это комментарий с важной информацией о варианте, добавленный пользователем. То, где она будет отображаться после добавления, описано здесь. Чтобы добавить или отредактировать интерпретацию варианта, нажмите на поле ввода, введите текст и кликните вне текстового поля для сохранения интерпретации:
В скобках рядом с названием раздела отобразится автор добавленной интерпретации - имя пользователя, который проинтерпретировал вариант:

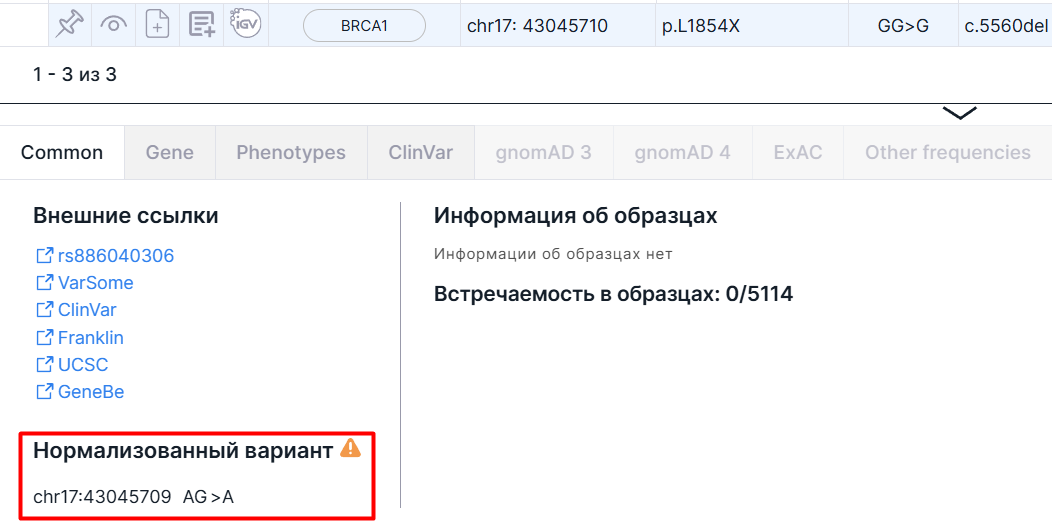
Нормализованный вариант#
У некоторых вариантов стартовая позиция, референсный аллель или альтернативный аллель, приведенные в загруженном VCF файле образца, могут не совпадать с левонормализованными. В таком случае в таблице SNV Viewer вариант будет записан в том же виде, что и в исходном VCF файле, а на панели детальной информации будут приведены нормализованные данные:
Информация о фазовой группе#
Фазовая группа - это объединение гомозиготных вариантов, расположенных в пределах определенного геномного интервала (окна размера N, который можно поменять, как описано здесь). Причем фазовая группа образовывается таким образом, чтобы хотя бы один вариант из группы оказывал эффект на белок-кодирующую часть хотя бы одного гена.
На панели детальной информации о фазовой группе будут приведены варианты, которые в неё входят (нажав на ссылки, можно открыть страницы детальной информации об этих вариантах):

На панели детальной информации о варианте, входящем в фазовые группы, будут отмечены эти группы (нажав на ссылки, можно открыть страницы детальной информации об этих группах):

Если вариант является частью фазовой группы с несовпадающей аминокислотной заменой, то
такой вариант будет отмечен значком ![]() .
Если вариант является частью фазовой группы с совпадающей аминокислотной заменой, то значка у варианта не будет.
.
Если вариант является частью фазовой группы с совпадающей аминокислотной заменой, то значка у варианта не будет.
Пример представления фазовых групп в IGV: в треке "phasing groups" располагаются фазовые группы. Слева в треке находится три фазовые группы: chr15:48505102 CA>TT (включает 2 варианта), chr15:48505102 CAA>TTG (включает 3 варианта), chr15:48505103 AA>TG (включает 2 варианта), по середине - одна фазовая группа chr15:48505109 CA>TG (включает 2 варианта) и справа - одна фазовая группа chr15:48505117 CA>AG (включает 2 варианта).

Gene#
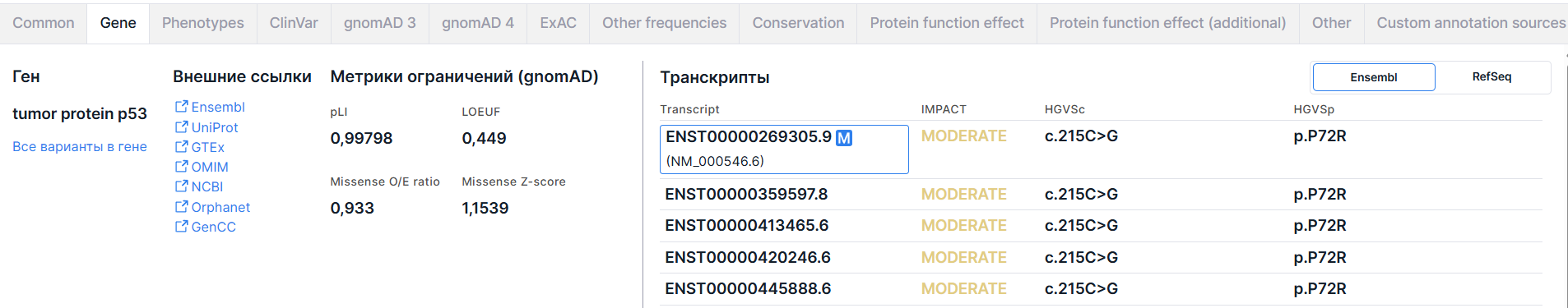
Вкладка содержит основную информацию о гене, включая метрики ограничений gnomAD, и о транскриптах, в которых располагается вариант.
1. Ген#
- Полное название гена. При наведении курсора на название гена можно увидеть, из каких баз данных было взято полное название:

- Поиск всех вариантов в гене: поиск всех вариантов, выявленных в данном образце и расположенных в этом же гене. Для поиска нажмите на ссылку "Все варианты в гене" - в соседней вкладке откроется страница SNV Viewer с фильтрацией по данному гену.
На заметку
При внесении любых изменений на открывшейся вкладке с поиском они сохранятся в SNV Viewer. В таком случае, если на изначальной странице SNV Viewer были применены фильтрация и/или сортировка вариантов, они могут сброситься или поменяться, а в SNV Viewer сохранится то состояние, которое получили на новой вкладке.
2. Внешние ссылки#
Ссылки на страницы с информацией о гене в различных базах данных: Ensembl, UniProt, GTEx, OMIM, NCBI, Orphanet, GenCC.
3. Метрики ограничений (gnomAD)#
Метрики генетических ограничений (constraint metrics) - метрики, которые отражают степень непереносимости геном функционально значимых мутаций и помогают оценить вероятность их патогенности:
- pLI (Probability of Loss-of-Function Intolerance) — вероятность того, что транскрипт чувствителен к вариантам потери функции (Lof; Loss of Function). Отражает вероятность принадлежности гена к категории гаплонедостаточных (haploinsufficient), то есть таких, в которых утрата одной копии приводит к функциональным последствиям. Рассчитывается на основе высоконадежных вариантов потери функции (pLoF; predicted Lof) по данным gnomAD. Диапазон значений: от 0 до 1. Гены с pLI >= 0.9 считаются непереносимыми к вариантам потери функции.
- LOEUF (Loss-of-function Observed/Expected Upper bound Fraction) — верхняя граница 90% доверительного интервала отношения наблюдаемых к ожидаемым высоконадёжным вариантам потери функции (pLoF). Более низкие значения (LOEUF < 0.35) указывают на более высокую степень генетического ограничения к потерям функции.
- Missense O/E ratio — верхняя граница 90% доверительного интервала отношения наблюдаемых (observed) к ожидаемым (expected) вариантам с заменой аминокислоты (missense variants; миссенс-варианты). Чем ниже значение, тем больше ген ограничен в отношении миссенс-вариантов.
- Missense Z-score — Z-оценка для миссенс-вариантов (вариантов с заменой аминокислоты) в транскрипте. Отражает, насколько число наблюдаемых миссенс-мутаций в данном гене отличается от ожидаемого количества, если бы мутации распределялись случайным образом по геному. Более высокие положительные значения Z-оценки (≥3.09) указывают на повышенную чувствительность гена к аминокислотным заменам, то есть на большую степень генетического ограничения к миссенс-вариантам. Более низкие или отрицательные значения Z-оценки, наоборот, указывают на то, что в гене встречается больше миссенс-мутаций, чем ожидалось, а значит, он терпим к таким изменениям.
4. Транскрипты#
На панели доступны транскрипты гена из двух баз данных: Ensembl и RefSeq. Модели транскриптов Ensembl аннотируются непосредственно на референсном геноме, а RefSeq аннотирует последовательности мРНК. Из-за различий в последовательностях между референсными геномами и отдельными мРНК некоторые из мРНК RefSeq могут не полностью сопоставляться с референсным геномом.
По умолчанию показана таблица с транскриптами из Ensembl. Чтобы перейти к таблице с транскриптами из RefSeq, нажмите на соответствующую кнопку переключателя:
![]()
На заметку
На странице "Настройки пользователя" можно выбрать, какая из моделей транскриптов генов будет показана по умолчанию.
Таблица транскриптов гена из базы Ensembl имеет следующие колонки:
- Transcript - идентификатор транскрипта из базы Ensembl
(ENSTxxxxxxxxxxx). Для первого, основного транскрипта также приведен транскрипт
из RefSeq (NM_xxxxxx.x).
Нажав на идентификаторы, можно открыть страницы транскриптов в этих базах.
Основной транскрипт гена, согласно MANE Select,
MANE plus clinical
или Ensembl canonical,
отмечен
 . Транскрипт гена,
выбранный основным для этого варианта, выделен рамкой. Значения Impact, HGVSc и HGVSp именно для этого
транскрипта приведены для варианта в таблице SNV Viewer.
. Транскрипт гена,
выбранный основным для этого варианта, выделен рамкой. Значения Impact, HGVSc и HGVSp именно для этого
транскрипта приведены для варианта в таблице SNV Viewer.
- IMPACT - предсказанное значение эффекта варианта на белок (см. таблицу со значениями эффектов варианта в аннотации). Значение эффекта HIGH раскрашено красным, MODERATE - жёлтым, а LOW и MODIFIER - серым.
- HGVSc - нуклеотидная замена в номенклатуре HGVS: префикс “c.” (coding; для замены в кодирующей последовательности ДНК) или “n.” (non-coding; для замены в некодирующей последовательности ДНК) + геномная позиция замещенного нуклеотида + референсный аллель > альтернативный аллель.
- HGVSp - аминокислотная замена в номенклатуре HGVS: префикс “p.” (protein) + референсная аминокислота + позиция аминокислоты в белке + новая аминокислота, получившаяся в результате замены.
Таблица транскриптов гена из базы RefSeq имеет следующие колонки:
- RefSeq Transcript - идентификатор транскрипта из базы RefSeq
(NM_xxxxxx.x). Основной транскрипт гена, согласно MANE Select,
MANE plus clinical
или Ensembl canonical,
отмечен . Транскрипт гена,
выбранный основным для этого варианта, выделен рамкой.
- HGVSc - нуклеотидная замена в номенклатуре HGVS: префикс “c.” (coding; для замены в кодирующей последовательности ДНК) или “n.” (non-coding; для замены в некодирующей последовательности ДНК) + геномная позиция замещенного нуклеотида + референсный аллель > альтернативный аллель.
- HGVSp - аминокислотная замена в номенклатуре HGVS: префикс “p.” (protein) + референсная аминокислота + позиция аминокислоты в белке + новая аминокислота, получившаяся в результате замены.
Phenotypes#
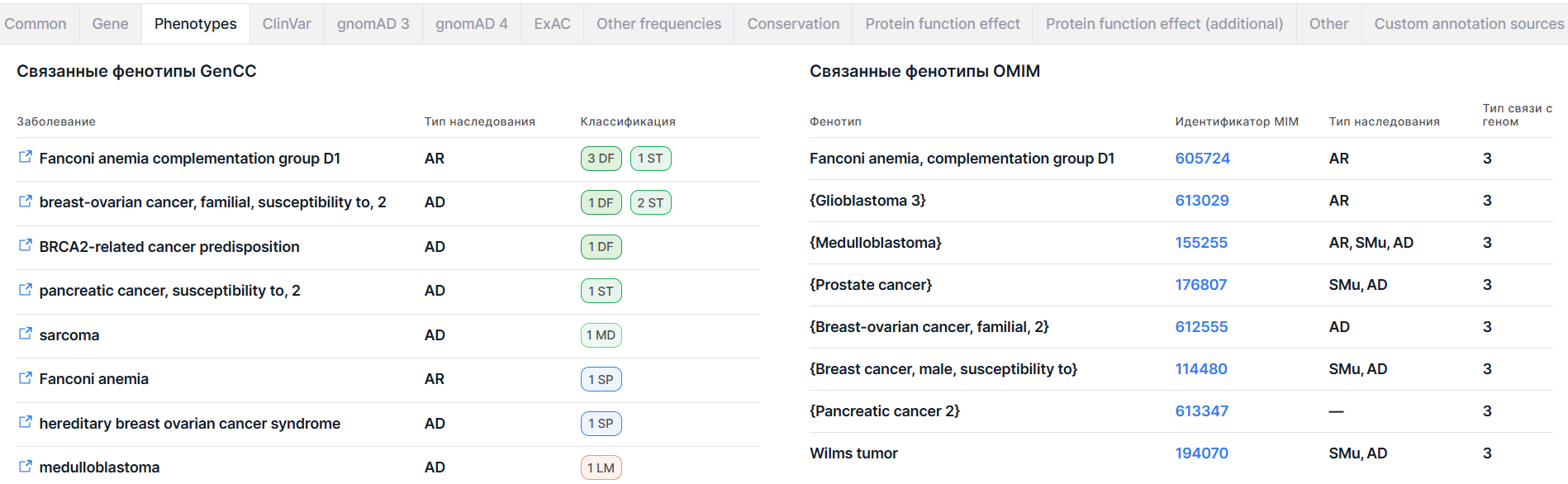
На вкладке приведена информация о фенотипах, ассоциированных с геном, в котором расположен вариант, по данным баз GenCC и OMIM. Информация о связанных фенотипах OMIM приводится, если вам предоставлен доступ к этой базе данных.
1. Связанные фенотипы GenCC#
Раздел содержит информацию о заболеваниях, ассоциированных с геном, в котором расположен вариант, по данным базы GenCC.
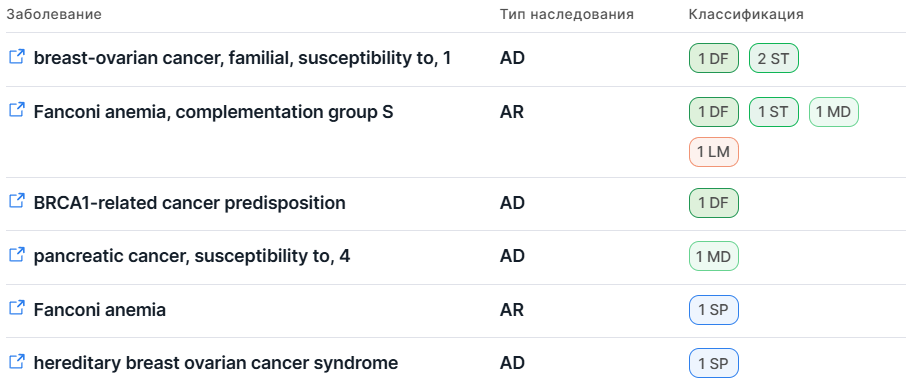
Информация представлена в виде таблицы со следующими колонками:
- Заболевание - полное название заболевания, связанного с геном. Для каждого заболевания
приводится ссылка на соответствующую запись в онтологии MONDO
(на сайте консорциума Monarch Initiative):
 .
При наведении курсора на значок ссылки отображается всплывающая подсказка с идентификатором заболевания в MONDO.
.
При наведении курсора на значок ссылки отображается всплывающая подсказка с идентификатором заболевания в MONDO. - Тип наследования заболевания:
- AD - Аутосомно-доминантный (Autosomal dominant);
- AR - Аутосомно-рецессивный (Autosomal recessive);
- MI - Митохондриальный (Mitochondrial);
- SD - Неполное доминирование (Semidominant);
- XL - X-сцепленный (X-linked);
- XLR - X-сцепленный рецессивный (X-linked recessive);
- YL - Y-сцепленный (Y-linked);
- — - Неизвестный (Unknown).
- Классификация - оценка связи гена и заболевания, основанная на силе доказательств, подтверждающих или опровергающих эту связь. Классификация отражает уровень клинической достоверности связи гена и заболевания.
Значения колонки представлены в виде значков с указанием количества источников, предоставивших доказательства соответствующего типа связи. При наведении курсора на значок отображается всплывающая подсказка, в которой указывается тип связи и перечисляются все источники, предоставившие соответствующие доказательства:

Типы связи гена и заболевания в GenCC:
 - Достоверная (Definitive);
- Достоверная (Definitive); - Сильная (Strong);
- Сильная (Strong); - Умеренная (Moderate);
- Умеренная (Moderate); - Поддерживающая (Supportive);
- Поддерживающая (Supportive); - Ограниченная (Limited);
- Ограниченная (Limited); - Спорные доказательства (Disputed Evidence);
- Спорные доказательства (Disputed Evidence); - Опровергнутые доказательства (Refuted Evidence);
- Опровергнутые доказательства (Refuted Evidence); - Связь с заболеванием не установлена (No Known Disease Relationship).
- Связь с заболеванием не установлена (No Known Disease Relationship).
Для каждого заболевания отображаются все типы связи, зарегистрированные для данного гена в GenCC. Значки классификации выводятся в порядке убывания силы доказательств (в соответствии с приведённым выше списком).
Таблица отсортирована по классификации связи гена и заболевания: сначала по убыванию силы доказательств, затем - по количеству источников, предоставивших доказательства соответствующего типа связи.
2. Связанные фенотипы OMIM#
Раздел содержит информацию о фенотипах, ассоциированных с геном, в котором расположен вариант, по данным базы OMIM. Доступ к данным из базы OMIM внутри Genomenal предоставляется при наличии у пользователя лицензии на доступ к базе.
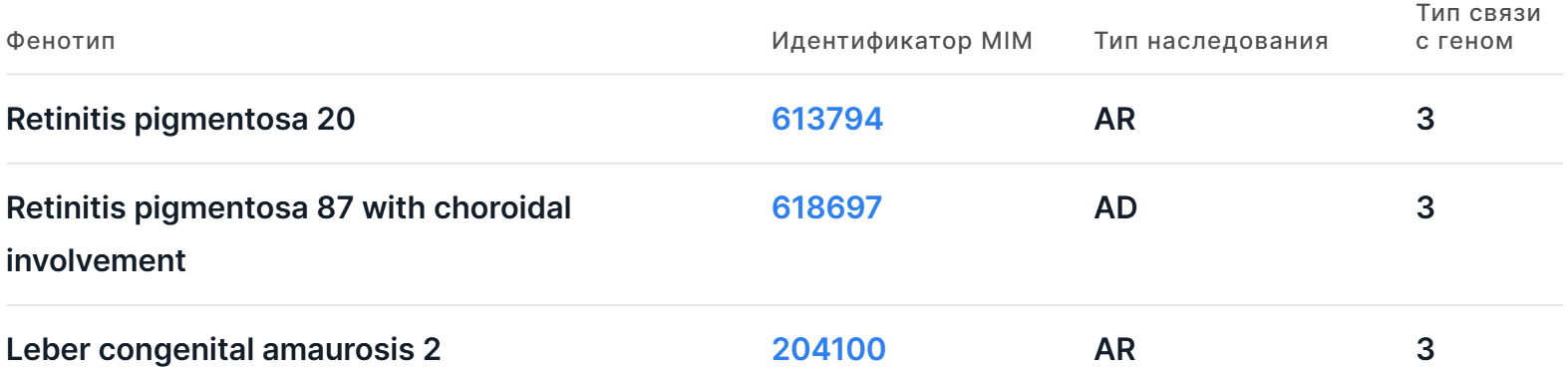
Информация представлена в виде таблицы со следующими колонками:
- Фенотип - полное название фенотипа, связанного с геном.
- Если перед названием фенотипа стоит знак вопроса (например, "?Bleeding disorder, platelet-type, 22"), это означает, что взаимосвязь между фенотипом и геном является предварительной.
- Если название фенотипа заключено в квадратные скобки (например, "[Urate oxidase deficiency]"), это указывает в основном на генетические вариации, которые приводят к явно аномальным значениям лабораторных тестов.
- Если название фенотипа заключено в фигурные скобки (например, "{Prostate cancer/brain cancer susceptibility, somatic}"), это обозначает мутации, которые способствуют предрасположенности к многофакторным заболеваниям или восприимчивости к инфекциям.
- Идентификатор MIM - уникальный идентификатор фенотипа в базе OMIM. Ссылка ведёт на страницу соответствующего фенотипа в OMIM.
- Тип наследования фенотипа:
- AD - Аутосомно-доминантный (Autosomal dominant);
- AR - Аутосомно-рецессивный (Autosomal recessive);
- DD - Дигенно-доминантный (Digenic dominant);
- DR - Дигенно-рецессивный (Digenic recessive);
- ICB - Унаследованный хромосомный дисбаланс (Inherited chromosomal imbalance);
- IC - Изолированные случаи (Isolated cases);
- Mi - Митохондриальный (Mitochondrial);
- Mu - Мультифакториальный (Multifactorial);
- SMo - Соматический мозаицизм (Somatic mosaicism);
- SMu - Соматическая мутация (Somatic mutation);
- XL - X-сцепленный (X-linked);
- XLD - X-сцепленный доминантный (X-linked dominant);
- XLR - X-сцепленный рецессивный (X-linked recessive);
- YL - Y-сцепленный (Y-linked);
- PD - Псевдоаутосомно-доминантный (Pseudoautosomal dominant);
- PR - Псевдоаутосомно-рецессивный (Pseudoautosomal recessive);
- — - Не указан (фенотип имеет пустое значение в колонке "Inheritance" в базе OMIM).
- Тип связи с геном - ключ карты фенотипа в OMIM, отображающей связи данного фенотипа с
ассоциированными фенотипами, группами родственных фенотипов (фенотипическими рядами) и генами:
1 - фенотип помещен на карту из-за его связи с геном, но лежащий в его основе дефект неизвестен;
2 - фенотип помещен на карту в результате использования статистических методов, мутация не обнаружена;
3 - молекулярная основа фенотипа известна, в гене была найдена мутация;
4 - синдром смежной дупликации или делеции генов, в котором участвуют несколько генов.
ClinVar#
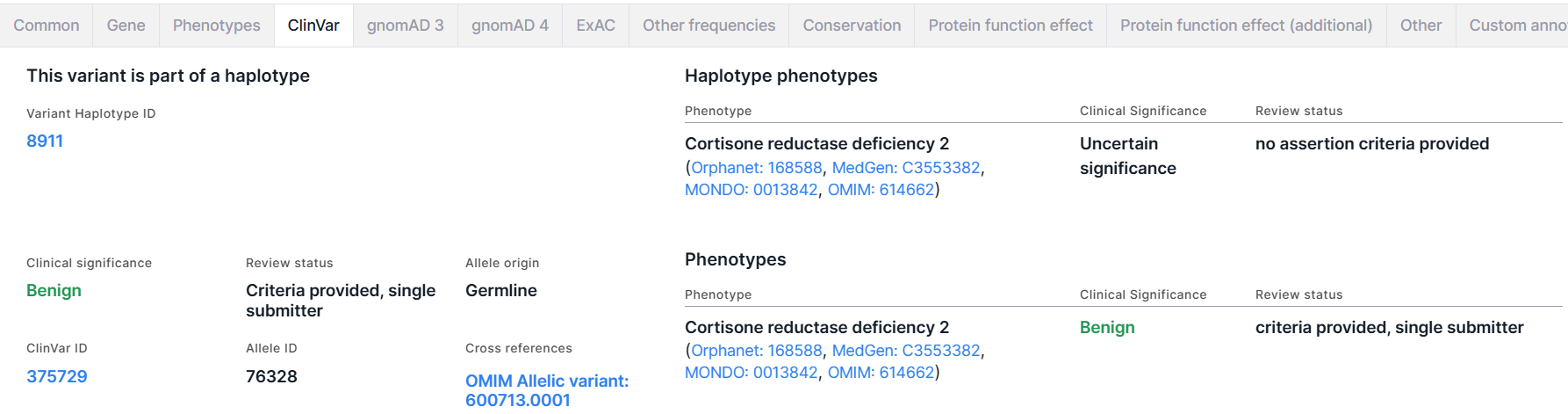
Вкладка содержит информацию о фенотипической значимости гаплотипа, если вариант является частью гаплотипа, и о фенотипической значимости варианта из базы данных ClinVar. В обоих разделах приводится таблица с фенотипами, которые наблюдались у людей с этим гаплотипом/вариантом и сообщались в ClinVar. Таблица имеет следующие колонки:
- Phenotype - название фенотипа (со ссылками на различные базы данных).
- Clinical Significance - клиническая значимость фенотипа (определение значений можно посмотреть тут). Клиническая значимость Benign раскрашена темно-зелёным; Likely benign - светло-зелёным; Likely pathogenic - оранжевым; Pathogenic - красным; все остальные значения, включая смешанные из разных клинических значимостей - чёрным.
- Review status - оценка источника данных, в котором была заявлена клиническая значимость фенотипа. Определение оценок можно посмотреть тут.
Haplotype phenotypes#
Раздел присутствует на вкладке, если вариант является частью гаплотипа. Раздел включает:
- Variant Haplotype ID - идентификатор гаплотипа, в который входит вариант. Перейдя по ссылке, вы попадете на страницу гаплотипа в ClinVar.
- Таблица с фенотипами, которые наблюдались у людей с этим гаплотипом и сообщались в ClinVar (см. описание колонок таблицы выше).
Phenotypes#
Раздел о фенотипической значимости варианта включает следующую информацию:
- Clinical significance - суммарная клиническая значимость всех фенотипов, которые наблюдались у людей с этим вариантом и сообщались в ClinVar. Определение значений можно посмотреть тут).
- ClinVar ID - идентификатор варианта в ClinVar. Перейдя по ссылке, вы попадете на страницу варианта в ClinVar.
- Review status - суммарная оценка всех источников данных, в которых была заявлена клиническая значимость ассоциированных с вариантом фенотипов. Определение оценок можно посмотреть тут.
- Allele ID - идентификатор аллеля варианта в ClinVar.
- Allele origin - суммарное происхождение аллеля варианта в разных источниках, предоставленных ClinVar.
- Cross references - ссылки на вариант в различных базах данных.
- Таблица с фенотипами, которые наблюдались у людей с этим вариантом и сообщались в ClinVar (см. описание колонок таблицы выше).
gnomAD 3#
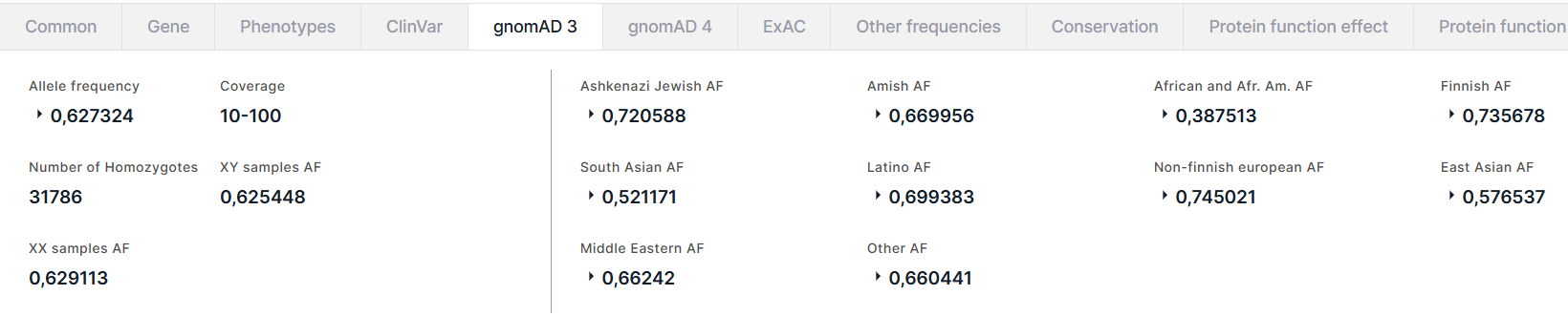
Вкладка содержит информацию об аллельных частотах варианта из базы данных gnomAD (версия 3). Слева на вкладке приведена общая информация о варианте:
- Allele frequency - суммарная частота альтернативного аллеля варианта в генотипах высокого качества. Для частоты приведены AC - количество альтернативного аллеля варианта в генотипах высокого качества, и AN - общее количество генотипов высокого качества.
- Coverage - средняя глубина покрытия по основаниям (диапазоны <10, 10-100, ⩾100).
- Number of Homozygotes - количество людей, гомозиготных по альтернативному аллелю, в популяции.
- XY samples AF - суммарная частота альтернативного аллеля в подпопуляции XY.
- XX samples AF - суммарная частота альтернативного аллеля в подпопуляции XX.
Справа на вкладке приведены частоты варианта в различных популяциях: Ashkenazi Jewish, Amish, African and African American (Afr. Am.), Finnish, South Asian, Latino, Non-finnish european, East Asian, Middle Eastern и Other (включает людей, которые не были однозначно сгруппированы с основными популяциями в анализе путем метода главных компонент). Для каждой популяции приведены суммарная частота альтернативного аллеля (AF) в генотипах высокого качества в популяции, частоты в подпопуляциях XY и XX и количество гомозигот в популяции:

gnomAD 4#
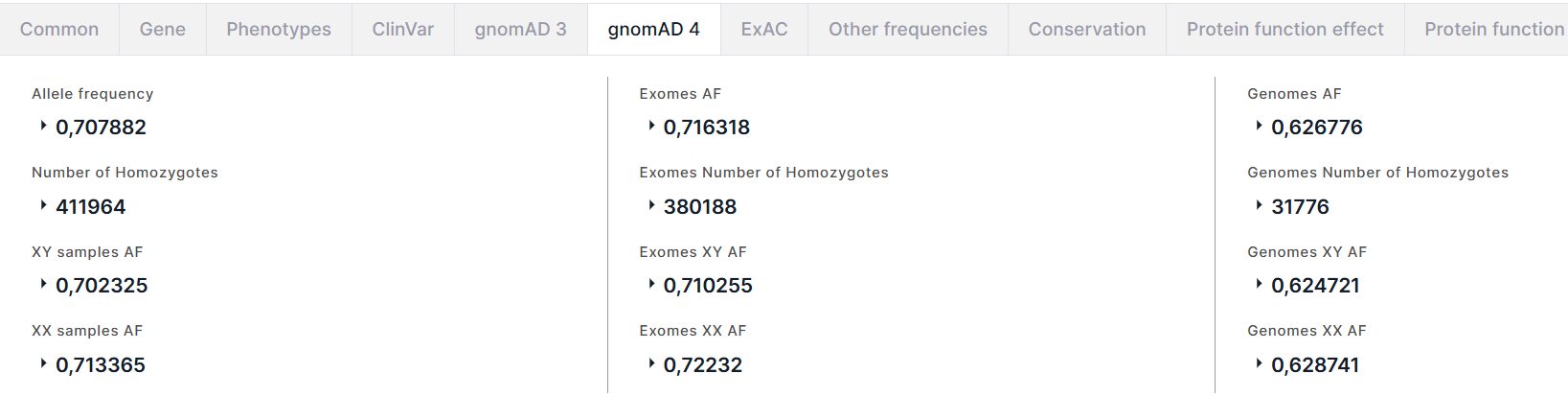
Вкладка содержит информацию об аллельных частотах варианта из базы данных gnomAD (версия 4). На вкладке представлены три столбца с информацией об аллельных частотах: слева - суммарная информация (в экзомных и геномных данных), по середине - только в экзомных данных, справа - только в геномных данных. Поля в этих столбцах идентичные:
- Allele frequency (AF) - суммарная частота альтернативного аллеля варианта в генотипах высокого качества. Для частоты приведены AC - количество альтернативного аллеля варианта в генотипах высокого качества, и AN - общее количество генотипов высокого качества.
- Number of Homozygotes - количество людей, гомозиготных по альтернативному аллелю, в популяции. Для количества гомозигот приведены XY samples и XX samples - количество людей, гомозиготных по альтернативному аллелю, в подпопуляции XY и подпопуляции XX, соответственно.
- XY samples AF (XY AF) - суммарная частота альтернативного аллеля в подпопуляции XY. Для частоты приведены XY samples AC (XY AC) - количество альтернативного аллеля варианта в генотипах высокого качества в подпопуляции XY, и XY samples AN (XY AN) - общее количество генотипов высокого качества в подпопуляции XY.
- XX samples AF (XX AF) - суммарная частота альтернативного аллеля в подпопуляции XX. Для частоты приведены XX samples AC (XX AC) - количество альтернативного аллеля варианта в генотипах высокого качества в подпопуляции XX, и XX samples AN (XX AN) - общее количество генотипов высокого качества в подпопуляции XX.
ExAC#
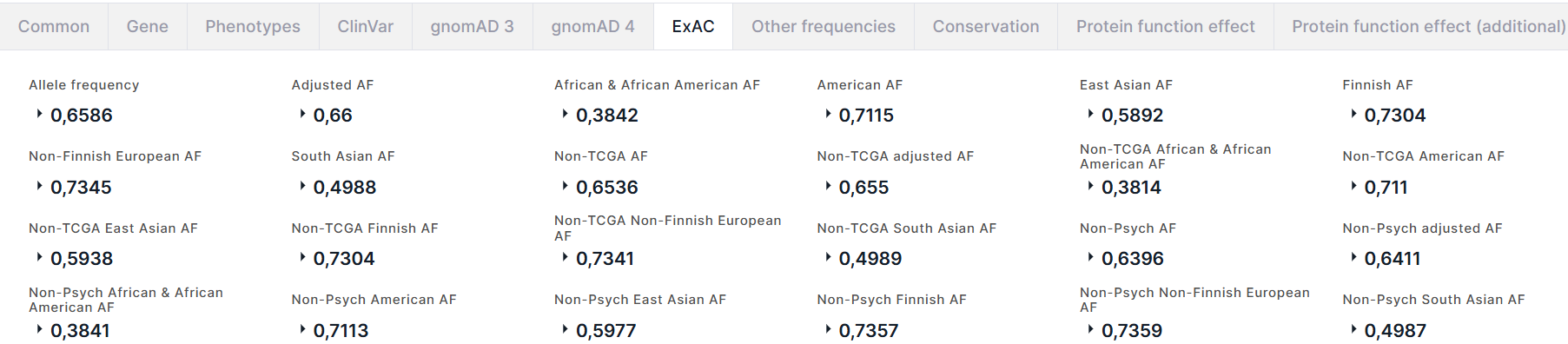
Вкладка содержит информацию об аллельных частотах варианта из базы данных ExAC:
- Allele frequency - суммарная частота альтернативного аллеля варианта.
- Adjusted AF - частота альтернативного аллеля в популяции с качеством генотипа ≥20 и глубиной ≥10.
- Частоты альтернативного аллеля в популяциях African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian.
- Частоты альтернативного аллеля в когортах non-TCGA (когорта, из которой были исключены образцы здоровой ткани онкобольных пациентов) и non-Psych (когорта, из которой были исключены образцы здоровой ткани пациентов, имеющих психические расстройства): суммарная частота альтернативного аллеля варианта в когорте (AF), частота аллеля в популяции когорты с качеством генотипа ≥20 и глубиной ≥10 (adjusted AF) и частоты аллеля в когорте в различных популяциях: African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian.
Для каждой частоты приведено количество альтернативного аллеля в генотипах высокого качества (Allele count):

Other frequencies#
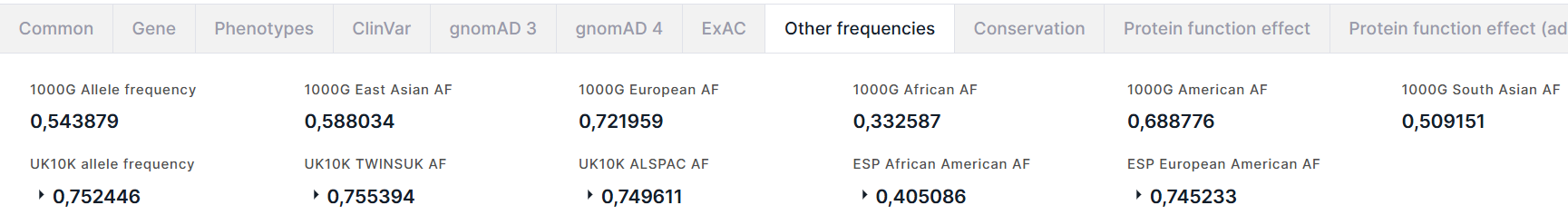
Вкладка содержит информацию об аллельных частотах варианта из проектов 1000 Genomes (1000G), UK10K и NHLBI GO Exome Sequencing Project (ESP).
Из проекта 1000 Genomes приведены:
- Allele frequency - суммарная частота альтернативного аллеля варианта;
- Частоты альтернативного аллеля в популяциях East Asian, European, African, American, South Asian.
Из проекта UK10K приведены:
- Allele frequency - суммарная частота альтернативного аллеля варианта в комбинированных генотипах в когорте UK10K (TwinsUK + ALSPAC);
- TWINSUK AF - частота альтернативного аллеля в TwinsUK - когорте, включающей взрослых близнецов, проживающих в Великобритании;
- ALSPAC AF - частота альтернативного аллеля в ALSPAC - когорте, включающей образцы тканей плодов.
Из проекта ESP приведены частоты альтернативного аллеля в популяциях African American и European American.
Для частот из проектов UK10K и ESP приведено количество альтернативного аллеля в генотипах высокого качества (Allele count):

Conservation#
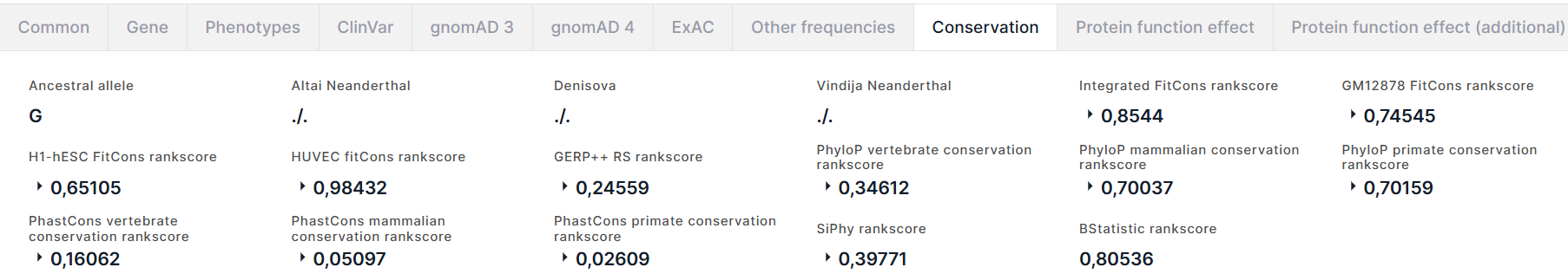
Вкладка содержит информацию о консервативности варианта:
- Ancestral allele - предковый аллель, полученный из множественных выравниваний полного генома Enredo-Pecan-Ortheus (EPO) с использованием вероятностного метода Ortheus. ACTG - аллели высокой достоверности, предковое состояние поддерживается двумя другими последовательностями; actg - аллели с низкой достоверностью, предковое состояние поддерживается только одной последовательностью; N - неудача, предковое состояние не поддерживается какой-либо другой последовательностью; - - современный вид содержит инсерцию в этом месте; . - нет покрытия в выравнивании.
- Altai Neanderthal - генотип глубоко секвенированного генома неандертальца Altai.
- Denisova - генотип глубоко секвенированного генома денисовского человека.
- Vindija Neanderthal - генотип глубоко секвенированного генома неандертальца Vindija.
- Оценки консервативности геномной позиции варианта, полученные с помощью метода fitCons. fitCons - вычислительный метод для оценки вероятности того, что точечная мутация в любой позиции в геноме повлияет на приспособленность. Объединяет функциональные анализы (например, ChIP-Seq) с селективным давлением, полученным с использованием метода INSIGHT. Результатом предсказания является оценка ρ в диапазоне [0-1], которая показывает долю геномных позиций, демонстрирующих конкретный паттерн результатов функционального анализа, которые находятся под селективным давлением. Из результатов fitCons приведены следующие оценки: Integrated FitCons rankscore - интегрированный показатель, полученный путем объединения информации из трех клеточных линий (HUVEC, H1 hESC и GM12878), и GM12878 FitCons rankscore, H1-hESC FitCons rankscore, HUVEC fitCons rankscore - оценки консервативности в клеточных линиях GM12878, H1 hESC и HUVEC, соответственно. Для каждого показателя, помимо rankscore, также приведены score и confidence value (значение достоверности оценки).
- Оценки консервативности геномной позиции варианта, полученные с помощью программы GERP++. GERP (Genomic Evolutionary Rate Profiling) идентифицирует высоко консервативные участки в множественных выравниваниях путем количественной оценки дефицитов замен. Эти дефициты представляют собой замены, которые произошли бы, если бы элемент был нейтральной ДНК, но не произошли, потому что элемент находился в функциональном ограничении. Авторы GERP называют такие дефициты "отклоненными заменами" (rejected substitutions, RS). Оценка "отклоненных замен" определяется количеством замен, ожидаемых при нейтральности ДНК, за вычетом замен, наблюдаемых в этой позиции. Из результатов оценки GERP++ приводятся rankscore (GERP++ RS rankscore), neutral rate (нейтральный уровень) и score.
- PhyloP vertebrate conservation rankscore, PhyloP mammalian conservation rankscore и PhyloP primate conservation rankscore и соответствующие score - оценки консервативности, полученные из выравнивания геномов позвоночных, млекопитающих и приматов, соответственно, программой PhyloP (phylogenetic P-values). Интерпретации оценок сравниваются с эволюцией, ожидаемой при нейтральном дрейфе.
- PhastCons vertebrate conservation rankscore, PhastCons mammalian conservation rankscore и PhastCons primate conservation rankscore и соответствующие scores - оценки консервативности, полученные из выравнивания геномов позвоночных, млекопитающих и приматов, соответственно, программой phastCons. PhastCons - это программа для выявления эволюционно консервативных элементов в множественном выравнивании с учетом филогенетического дерева. Основана на филогенетической скрытой марковской модели.
- SiPhy rankscore и score - оценки консервативности геномной позиции варианта, полученные программой SiPhy, используя простое обобщение оценки паттерна замены.
- BStatistic rankscore - оценка консервативности геномной позиции варианта, полученная из анализа геномного распределения полиморфизмов человека и различий в последовательностях среди пяти видов приматов относительно расположения паттернов консервативных последовательностей (подробнее можно прочитать здесь).
Protein function effect#
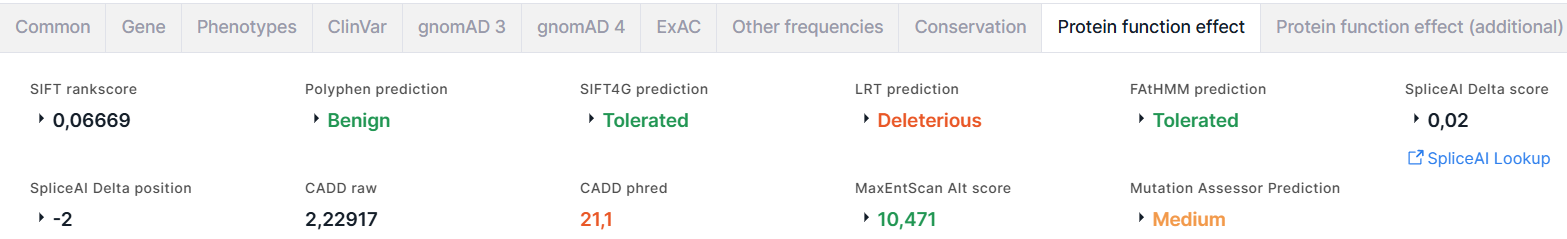
Вкладка содержит результаты предсказания эффекта аминокислотной замены на функцию белка, полученные с помощью следующих алгоритмов и программ:
- SIFT (Sorting Intolerant From Tolerant) предсказывает, влияет ли аминокислотная замена на функцию белка, основываясь на гомологии последовательностей и физических свойствах аминокислот. Результатом предсказания являются rankscore и score, значения которых варьируют от 0 до 1. Аминокислотная замена считается повреждающей, если score ≤ 0,05, и толерантной, если score > 0,05.
- PolyPhen (Polymorphism Phenotyping) предсказывает возможное влияние
аминокислотной замены на структуру и функции белка, используя восемь свойств белковой последовательности и три
свойства структуры белка. Результатом предсказания являются:
1. prediction: Benign (доброкачественный) раскрашен зелёным; Possibly damaging (возможно повреждающий) - оранжевым; Probably damaging (вероятно повреждающий) - красным;
2. score, значения которого варьируют от 0 до 1: чем ниже, тем более вероятно, что вариант является доброкачественным, чем выше - повреждающим. - SIFT4G (SIFT Databases for Genomes) - более быстрая версия SIFT. Результатом предсказания являются score, rankscore, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- LRT (Likelihood Ratio Test) идентифицирует повреждающие мутации, которые нарушают высококонсервативные аминокислоты в белок-кодирующих последовательностях, основываясь на эволюционной модели последовательности ДНК. Результатом предсказания являются score, rankscore, omega, prediction: Neutral (нейтральный) раскрашен зелёным; Deleterious (повреждающий) - красным.
- FAtHMM (Functional Analysis through Hidden Markov Models) прогнозирует функциональные последствия как белок-кодирующих вариантов, так и некодирующих вариантов, основываясь на гомологии последовательностей. Результатом предсказания являются score, rankscore, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- SpliceAI - глубокая остаточная нейронная сеть, которая
предсказывает, является ли каждая позиция в транскрипте пре-мРНК донором сплайсинга, акцептором сплайсинга или ни тем,
ни другим, используя в качестве входных данных только геномную последовательность транскрипта пре-мРНК. Результатом
вычисления вероятности влияния вариантов на сайты сплайсинга являются:
1. Delta score (DS) - вероятность того, что вариант влияет на сплайсинг в любой позиции в пределах заданного интервала вокруг него (+/- 50 п.н. по умолчанию). Оценки дельты варьируются от 0 до 1:
- Acceptor gain - вероятность того, что позиция в пределах заданного интервала вокруг варианта (+/- 50 п.н. по умолчанию) используется как акцептор сплайсинга;
- Acceptor loss - вероятность того, что позиция в пределах заданного интервала вокруг варианта (+/- 50 п.н. по умолчанию) не используется как акцептор сплайсинга;
- Donor gain - вероятность того, что позиция в пределах заданного интервала вокруг варианта (+/- 50 п.н. по умолчанию) используется как донор сплайсинга;
- Donor loss - вероятность того, что позиция в пределах заданного интервала вокруг варианта (+/- 50 п.н. по умолчанию) не используется как донор сплайсинга.
За DS принимается максимальное значение среди этих четырех показателей. Значения 0 ≤ DS < 0.2 раскрашены чёрным, 0.2 ≤ DS < 0.5 - зелёным, 0.5 ≤ DS < 0.8 - оранжевым, 0.8 ≤ DS ≤ 1 - красным.
2. Delta position - позиция (в п.н.) относительно варианта, в которой изменяется сплайсинг (положительные значения — ниже позиции варианта, отрицательные значения — выше). Delta position соответствует значению позиции, полученной для показателя (Acceptor gain, Acceptor loss, Donor gain или Donor loss) с максимальным значением DS среди остальных.
3. Ссылка на сайт SpliceAI Lookup, где приведена более расширенная информация об оценках для варианта. - CADD (Combined Annotation-Dependent Depletion) оценивает вредоносность
вариантов, интегрируя несколько аннотаций варианта в одну метрику путём сопоставления вариантов, оставшихся после
естественного отбора, со смоделированными вариантами. Результатом оценки являются:
- Raw - необработанная оценка, относительное значение. Интерпретируется как степень, в которой профиль аннотации для данного варианта предполагает, что вариант, вероятно, будет «наблюдаемым» (отрицательные значения) по сравнению с «моделированным» (положительные значения). Более высокие значения оценки указывают на то, что вариант с большей вероятностью будет иметь вредоносные последствия.
- Phred - Phred-масштабированная нормализованная оценка. Значения Phred < 10 раскрашены зелёным,
10 ≤ Phred < 20 - оранжевым, Phred ≥ 20 - красным. - MaxEntScan предсказывает пригодность сайта варианта для сплайсинга на основе модели максимальной энтропии. Результатом являются оценки для альтернативного и референсного нуклеотидов варианта, предсказывающие потерю естественного сайта сплайсинга: Alt score и Ref score, и разница между этими оценками: Diff. Значения Alt score < -19 раскрашены красным, -19 ≤ Alt score < -3 - оранжевым, Alt score ≥ -3 - зелёным.
- MutationAssessor предсказывает функциональное влияние аминокислотной
замены варианта на основе эволюционной консервативности затронутой аминокислоты в белковых гомологах.
Результатом оценки являются:
1. Prediction - предсказание функциональности влияния замены: функциональное: High (высокое) раскрашено красным, Medium (среднее) - оранжевым, или нефункциональное: Low (низкое) и Neutral (нейтральное) раскрашены зелёным.
2. Score - численное значение предсказания. Значения Score ≥ 3,5 соответствуют высокому влиянию,
3,5 < Score ≤ 1,935 - среднему, 1,935 < Score ≤ 0,8 - низкому, 0,8 < Score - нейтральному.
Дополнительные результаты предсказания эффекта аминокислотной замены варианта на функцию белка расположены на соседней вкладке Protein function effect (additional):
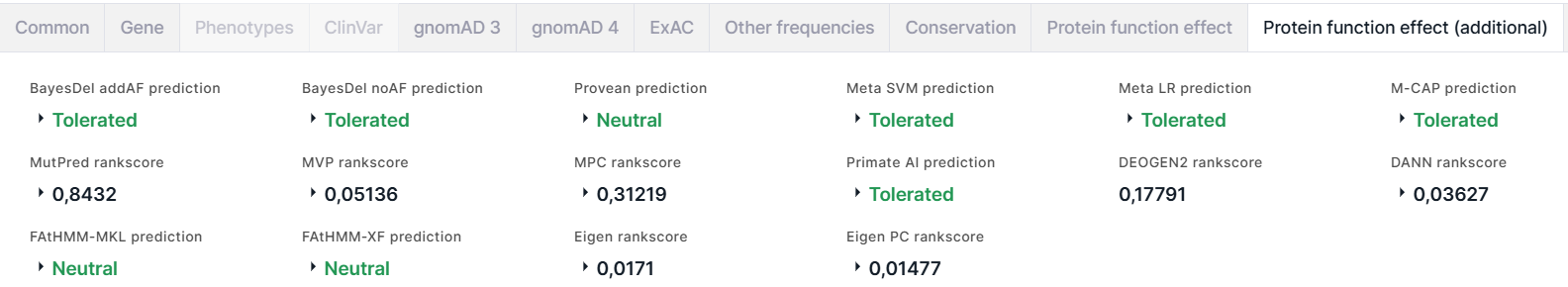
- BayesDel - это мета-оценка вредоносности вариантов. Работает для кодирующих
и некодирующих вариантов; однонуклеотидных вариантов и небольших вставок/делеций. Чем выше оценка, тем больше
вероятность того, что вариант является патогенным. Результатом предсказания
являются:
1. addAF prediction - предсказание вредоносности вариантов с учётом MaxAF (максимальной частоты аллеля среди популяций): Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным. Значению предсказания соответствуют оценки addAF score и addAF rankscore, на основе которых было сделано предсказание. Порог между оценками, соответствующими повреждающему и толерантному значению предсказания, составляет 0,0692655.
2. noAF prediction - предсказание вредоносности вариантов без учёта MaxAF: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным. Значению предсказания соответствуют оценки noAF score и noAF rankscore, на основе которых было сделано предсказание. Порог между оценками, соответствующими повреждающему и толерантному значению предсказания, составляет -0,0570105. - PROVEAN (Protein Variation Effect Analyzer) предсказывает, влияет ли аминокислотная замена на биологическую функцию белка. Результатом предсказания являются score, rankscore, prediction: Neutral (нейтральный) раскрашен зелёным; Damaging (повреждающий) - красным.
- Meta SVM (Support Vector Machine) - мета-аналитическая структура, использующая метод опорных векторов и интегрирующая 10 скоров (SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, GERP++, MutationTaster, MutationAssessor, FATHMM, LRT, SiPhy, PhyloP) и максимальную частоту, наблюдаемую в популяциях 1000 геномов. Результатом вычислений являются score, rankscore, reliability index, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- Meta LR (Logistic Regression) - мета-аналитическая структура, использующая логистическую регрессию и интегрирующая 10 скоров (SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, GERP++, MutationTaster, MutationAssessor, FATHMM, LRT, SiPhy, PhyloP) и максимальную частоту, наблюдаемую в популяциях 1000 геномов. Результатом вычислений являются score, rankscore, reliability index, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- M-CAP (Mendelian Clinically Applicable Pathogenicity) - классификатор патогенности редких миссенс-мутаций в геноме человека, настроенный на высокую чувствительность, необходимую в клинике, комбинируя различные оценки патогенности (включая SIFT, Polyphen-2 и CADD) с новыми функциями и мощной моделью. Результатом предсказания являются score, rankscore, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- MutPred - классификатор аминокислотных замен как ассоциированных с заболеванием или нейтральных, предсказывает молекулярную причину заболевания. Результатом предсказания являются rankscore, score, transcript ID (идентификатор транскрипта из базы UniProtKB, для которого было сделано предсказание эффекта аминокислотной замены на функцию белка), amino acid change (аминокислотная замена, для которой был предсказан эффект на функцию белка), top 5 features (основные пять эффектов замены на функцию белка). Чем больше значение скора, тем более вероятно, что вариант будет иметь повреждающий эффект.
- MVP (Missense Variant Pathogenicity) - метод прогнозирования, который использует глубокую остаточную сеть для объединения больших наборов обучающих данных и множества коррелированных предсказателей. Результатом предсказания являются rankscore и score.
- MPC (Missense badness, PolyPhen-2, and Constraint) - метрика повреждающих de novo миссенс-мутаций, основанная на идентификации субгенных областей, в которых отсутствует миссенс-вариация. Результатом предсказания являются rankscore и score. Чем больше значение скора, тем более вероятно, что вариант патогенный.
- Primate AI - это глубокая остаточная нейронная сеть для классификации патогенности миссенс-мутаций, основанная на распространенных вариантах приматов, отличных от человеческих. Результатом предсказания являются score, rankscore, prediction: Tolerated (толерантный) раскрашен зелёным; Damaging (повреждающий) - красным.
- DEOGEN2 - метод, предсказывающий, влияет ли вариант, кодирующий белок, на здоровье человека-носителя. Метод включает разнородную информацию о молекулярных эффектах вариантов, задействованных доменах, релевантности гена и взаимодействиях, в которых он принимает участие. Результатом предсказания является rankscore.
- DANN (Domain-Adversarial Neural Network) - глубокая нейронная сеть, переобученная на основе данных обучения CADD. Результатом вычислений являются rankscore и score. Чем больше значение скора, тем более вероятен повреждающий эффект варианта.
- FAtHMM-MKL (Multiple Kernel Learning) - метод машинного обучения, объединяющий функциональные аннотации из ENCODE со скрытыми марковскими моделями на основе нуклеотидов. Результатом предсказания являются p-value, rankscore, group of features, prediction: Neutral (нейтральный) раскрашен зелёным; Damaging (повреждающий) - красным.
- FAtHMM-XF (eXtended Feature) - метод прогнозирования патогенных точечных мутаций в геноме человека, который опирается на обширный набор функций и особенно хорошо предсказывает в некодирующих областях. Результатом предсказания являются p-value (чем меньше значение, тем больше вероятность того, что вариант оказывает повреждающий эффект), rankscore, prediction: Neutral (нейтральный) раскрашен зелёным; Damaging (повреждающий) - красным.
- Eigen - спектральный подход к функциональной аннотации генетических вариантов в кодирующих и некодирующих областях. Результатом аннотации являются rankscore и score.
- Eigen-PC - более простой мета-скор, который основан на собственном разложении ковариационной матрицы аннотаций и использовании главного собственного вектора для взвешивания отдельных аннотаций. Результатом оценки являются rankscore, score и score in phred scale.
Other#
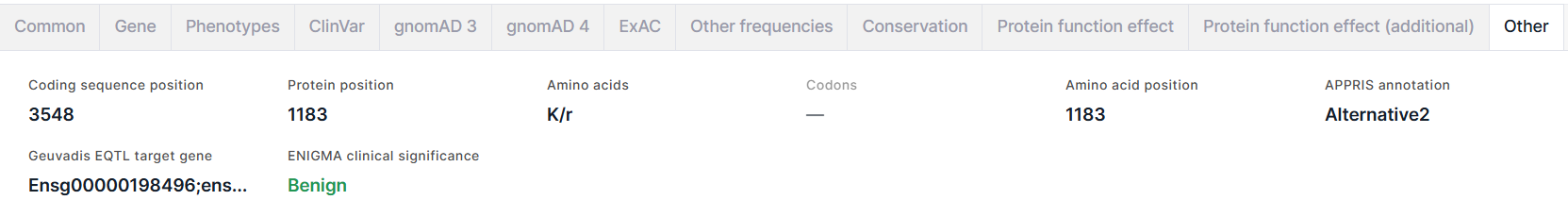
Влкадка содержит дополнительную информацию о варианте:
- Coding sequence position - геномная позиция референсного аллеля варианта.
- Protein position - позиция референсной аминокислоты в белке.
- Amino acids - референсная аминокислота/новая аминокислота, получившаяся в результате замены.
- Codons - аминокислотная замена, записанная в виде кодонов. Пример: GAG/CAG = E/Q (глутоминовая кислота -> глутамин).
- Amino acid position - позиция референсной аминокислоты в белке.
- APPRIS annotation - аннотация изоформ сплайсинга человека. Для каждого гена изоформа выбирается основной путем объединения информации о структуре белка, функционально важных остатков и данных межвидового выравнивания. Возможные значения аннотации: PRINCIPAL1-5 (основные изоформы, где 1 - наиболее надежная), ALTERNATIVE1 (модель кандидатного транскрипта, являющаяся консервативной как минимум у трех протестированных видов не из отряда приматов), ALTERNATIVE2 (модель кандидатного транскрипта, являющаяся консервативной менее чем у трех протестированных видов не из отряда приматов).
- Geuvadis EQTL target gene - идентификаторы таргетных генов из Ensembl, выявленных путем анализа локусов количественных признаков экспрессии (eQTL) в проекте GEUVADIS (Genetic European Variation in Disease).
- ENIGMA clinical significance - клиническая значимость варианта, если он расположен в известном или предполагаемом гене предрасположенности к раку молочной железы и/или яичников (BRCA1, BRCA2 и др.), выявленная экспертами из консорциума ENIGMA. Клиническая значимость Benign раскрашена темно-зелёным; Likely benign - светло-зелёным; Likely pathogenic - оранжевым; Pathogenic - красным; все остальные значения, включая смешанные из разных клинических значимостей - чёрным. При наведении курсора на значение поля вы увидите комментарий о клинической значимости.
Custom annotation sources#
Вкладка есть на панели детальной информации, если хотя бы одна пользовательская аннотация была добавлена в систему до того, как варианты в образце были проаннотированы. Вкладка содержит значения колонок из пользовательских аннотаций, которыми был проаннотирован вариант:

Данные удаленной аннотации помечены как "deleted" для вариантов, которые были аннотированы с её помощью до её удаления.