Variant Pathogenicity
The variant pathogenicity can be determined either manually (by user), by the ACMG classification, or by
the pathogenicity database uploaded as a custom annotation. Information about the variant pathogenicity is located
in "Pathogenicity" column in SNV Viewer. If the pathogenicity was not determined, then the column value
will be ![]() (not applicable).
(not applicable).
Manually determined pathogenicity#
To determine the variant pathogenicity by yourself, click on the pathogenicity
button ![]() in the variant row
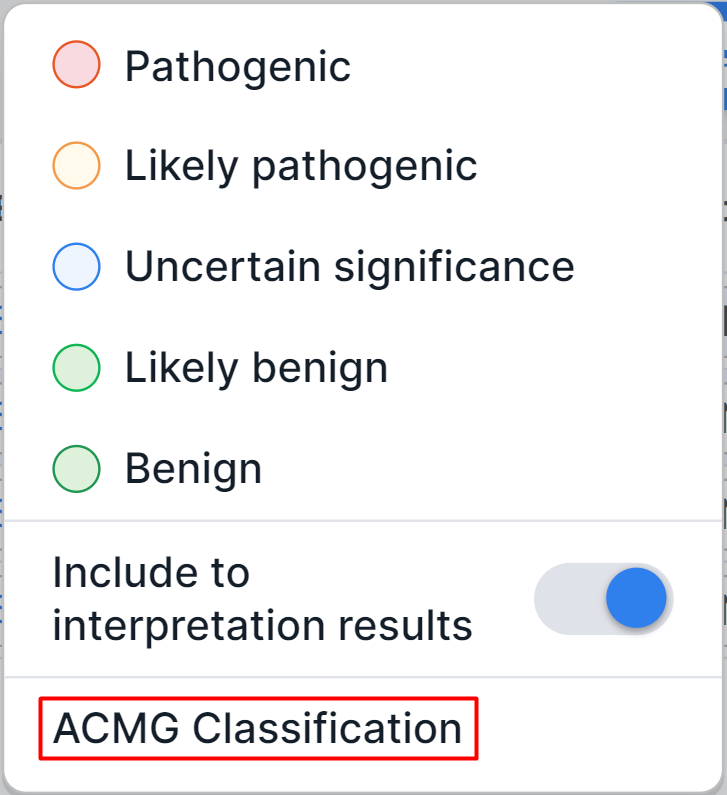
("Pathogenicity" column) and select the required option in the dialog:
in the variant row
("Pathogenicity" column) and select the required option in the dialog:

All variants with a certain pathogenicity class are automatically added to the sample interpretation results. If you want to determine the pathogenicity class of a variant but not include it to the interpretation results, disable the "Include to interpretation results" option in the same dialog.
info
If a variant has been determined as Pathogenic or Likely pathogenic, it is automatically included to report (if pathogenicity has been confirmed).
Pathogenicity can be overdetermined in the same dialog.
You can also determine pathogenicity in "ACMG Classification" tab using the ACMG criteria, as described here.
info
The variant pathogenicity class determined manually is stored in the system and will automatically be determined for the same variant in samples annotated later with a warning to confirm the pathogenicity.
ACMG Classification#
"ACMG Classification" is a tab on the variant details page
that provides the variant interpretation criteria developed
by American College of Medical Genetics and Genomics (ACMG). To get to the tab,
click on the pathogenicity button ![]() in
the variant row ("Pathogenicity" column) and click on the "ACMG Classification" option in the dialog:
in
the variant row ("Pathogenicity" column) and click on the "ACMG Classification" option in the dialog:
You will see "ACMG classification" tab on the variant details page (you can also access the tab from other tabs on the page).
On the left side of the tab, there is a navigation bar for ACMG criteria grouped by category. When you click on the name of a certain criteria group, it will be displayed on the right side of the page, and when you click on a certain criterion, you will see the corresponding group criterion. Criteria that are undefined for the variant are shown in grey; criteria that does not meet the variant - in black; benign criteria that meet the variant - in green; and pathogenic criteria that meet the variant - in orange.

On the right side of the tab, there is a detailed information about each criterion by group. For each criterion, there are:
- Criterion's code and significance presented as an icon. The icon color matches the color on
the navigation bar on the left: grey means that the criterion is
undefined for the variant; black - the criterion doesn't meet the
variant;
green - the benign criterion that meets the variant; orange - the pathogenic criterion that meets the variant. - Decision: Automatic (the criterion state is determined automatically), UNDEFINED (the criterion state
is undefined) or MANUALLY CHANGED (the criterion state was determined by the user; to return to automatic
determination of the criterion's state and significance,
click on
 );
); - Criterion state: UNDEFINED - the criterion is not defined for the variant; UNMET - the criterion does not meet the variant; MET - the criterion meets the variant. The selected state is indicated in blue. The state of the criterion can be changed by clicking on the button of the required state.

- The criterion can be expanded by clicking on
 .
Here you can add a comment to the criterion and change the criterion significance level: supporting,
moderate (if if there is no "Stand alone" option), strong; very strong (if if there is no "Stand alone" option);
stand alone (if there are no options "Moderate" and "Very strong"; independent criterion of very high significance).
There will also be "Literature" section with a list of publications associated with the criterion, if such
associations were
added on "Literature" tab.
.
Here you can add a comment to the criterion and change the criterion significance level: supporting,
moderate (if if there is no "Stand alone" option), strong; very strong (if if there is no "Stand alone" option);
stand alone (if there are no options "Moderate" and "Very strong"; independent criterion of very high significance).
There will also be "Literature" section with a list of publications associated with the criterion, if such
associations were
added on "Literature" tab.
1. Population data#
At the top of the section, there are:
- gnomAD 3 AF is the total allele frequency in gnomAD v3 database;
- ExAC AF is the total allele frequency in ExAC database;
- 1000G AF is the total allele frequency in the 1000 Genomes Project;
- Max AF is the maximum allele frequency among all frequencies in different populations according to different databases;
- Min AF is the minimum allele frequency among all frequencies in different populations according to different databases.
Population data criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Pathogenic Moderate 2 (PM2) | Moderate | Pathogenic | Absent from controls (or at extremely low frequency if recessive: ≤0.01% for autosomal dominant diseases or ≤0.5% for autosomal recessive diseases) in gnomAD, 1000 Genomes or ExAC. Must be covered to a sufficient read depth/quality (>100) in database. |
| Stand-alone evidence of benign impact 1 (BA1) | Very strong; independent criterion | Benign | The population allele frequency is above 5% in gnomAD, 1000 Genomes, or ExAC and the variant is not on the exception list by ClinGen SVI. |
| Strong evidence of benign 1 (BS1) | Strong | Benign | Allele frequency is above 1% and is greater than expected for disorder: 1. Variants known to be pathogenic for dominant disorders should have allele frequencies in the general population below the disease incidence. 2. Variant known to be pathogenic for recessive disorders should have heterozygous frequencies consistent with their disease incidence. - For an autosomal dominant disorder with high penetrance it is acceptable to use this criterion as stand-alone evidence to classify a variant as likely benign. - Be cautious using this code based on a low number of alleles in gnomAD if the disease is late onset or has variable penetrance/expressivity. - To avoid duplication, the criterion is applied as UNMET if the variant meets the BA1 or PM2 criteria. |
| Strong evidence of benign 2 (BS2) | Strong | Benign | Observed in a healthy adult individual for a (1) recessive (homozygous), (2) dominant (heterozygous), or (3) X-linked (hemizygous) disorder with full penetrance expected at an early age. - Use the criterion if it is reasonable to believe that individuals with the variant in large population databases are truly unaffected. The evidence can be accessed from gnomAD, the literature, or in-house data. For example, gnomAD shows the age distribution of heterozygous or homozygous variant carriers. - Be cautious using this criterion based on a low numbers if the disease is late-onset or has variable penetrance/expressivity. |
2. Odds ratio#
Odds ratio criterion#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion | Additional options |
| Pathogenic Strong 4 (PS4) | Strong | Pathogenic | The prevalence of the variant in affected individuals is significantly higher than in controls. Relative risk or odds ratio, as obtained from case-control studies, is >5.0, and the confidence interval around the estimate does not include 1.0. - Case-control study data is rarely available and do not reach statistical significance for very rare variants and rare diseases, therefore use the criterion: 1. at moderate level if the same variant has been previously identified in multiple (two or more) unrelated affected individuals and has not been reported in gnomAD (or at extremely low frequency if autosomal recessive); 2. at supporting level if the same variant has been previously identified in one unrelated affected individual and has not been reported in gnomAD (or at extremely low frequency if autosomal recessive); 3. at supporting level if the same variant has been previously identified in multiple unrelated individuals but is not absent in controls. - The patient phenotype must be consistent with the known disease spectrum. - In practice this is most applicable to autosomal dominant disorders (for autosomal recessive disorders use PM3). - Both PS4 and PM2 can be used together. | To open a variant in GWAS Catalog for human genome-wide associations, expand the criterion description by clicking on  . . |
3. Uniq Pheno#
Uniq Pheno criterion#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Supporting evidence of pathogenicity 4 (PP4) | Supporting | Pathogenic | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology. Before using the criterion, it is essential to make sure that: 1. all the known genes associated with the disorder have been analysed using a highly sensitive method appropriate for the reported types of variants; 2. variants in these known genes explain the majority of cases with that clinical diagnosis. |
4. Cosegregation#
At the top of the section, you can calculate the logarithm of the odds (LOD) score for a certain type of segregation. LOD scores calculated in different families with the same variant can be added to assign a final score. The final LOD score can then be used to determine the criterion significance level. To calculate LOD score, do the following:
- Select segregation type: dominant/X-linked or recessive;
- Enter the values of the number of affected segregations and unaffected segregations (if the recessive segregation was chosen) (natural numbers);
- Click outside the input box.
The LOD score for dominant/X-linked segregation is calculated by the formula:
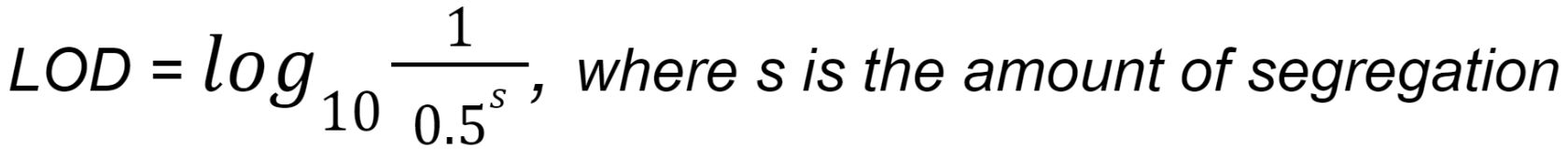
The LOD score for recessive segregation is calculated by the formula:

Cosegregation criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Supporting evidence of pathogenicity 1 (PP1) | Supporting | Pathogenic | Located in a gene definitively known to cause the disease and that has a co-segregation with this disease in multiple affected family members. - Before using the criterion, determine the segregation of a variant with disease phenotypes in pedigrees with affected patients. Use the criterion with an appropriate significance level if a variant was found to co-segregate with multiple affected family members. Use BS4 if there is a lack of segregation of a variant with affected members. - Based on the recommendations from ClinGen The Hearing Loss Working Group, use the criterion: 1. at supporting level if LOD = 0.6; 2. at moderate level if LOD = 1.2; 3. at strong level if LOD = 1.5. |
| Strong evidence of benign 4 (BS4) | Strong | Benign | Located in a gene that has lack of segregation with the disease in affected family members. - Before using the criterion, determine the segregation of a variant with disease phenotypes in pedigrees with affected patients. Use the criterion if there is a lack of segregation of a variant with affected members. Use PP1 with an appropriate significance level if a variant was found to co-segregate with multiple affected family members. |
5. De novo Variants#
The Sequence Variant Interpretation (SVI) Working Group proposes a point-based system to determine the strength of de novo evidence (PS2 and PM6 criteria) based upon three parameters:
- choose PS2 if de novo status confirmed and PM6 if not;
- phenotypic consistency;
- number of de novo observations.
De novo Variants criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Pathogenic Strong 2 (PS2) | Strong | Pathogenic | De novo in a patient with the disease and no family history. Maternity and paternity confirmation is required. |
| Pathogenic Moderate 6 (PM6) | Moderate | Pathogenic | Assumed de novo, but without confirmation of paternity and maternity. |
For De novo Variants criteria, you can add additional unrelated patients (probands) with the same phenotype:
- Expand the criterion description by clicking on ;
- For Proband 1 (the name can be changed by clicking on the current one and entering a new one), click on the search box and select the appropriate consistency between phenotype and gene;
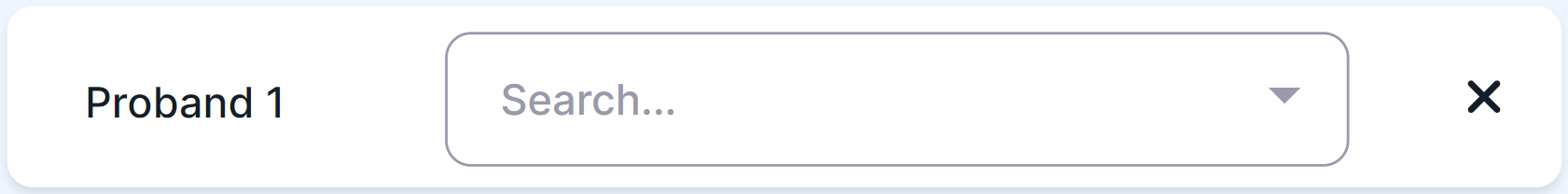
- To add another patient, click on
 .
.
After adding all patients, click on the button that appears to apply the criterion significance level corresponding to the phenotypes of the patients:

After that, the criterion state will change to MET (the criterion meets the variant) and the criterion significance level will correspond to the phenotypes of the added patients.
To delete a patient, click on ![]() .
.
6. Cis-trans#
Cis-trans criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion | Additional options |
| Pathogenic Moderate 3 (PM3) | Moderate | Pathogenic | Detected in trans with a pathogenic variant for recessive disorders. - The Sequence Variant Interpretation (SVI) Working Group proposes a point-based system to determine the criterion significance level for variants in trans observations based upon variant phasing and classification of the variant occurring on the other allele. - SVI recommends a revision to the criterion definition: "for recessive disorders, detected in trans with a pathogenic or likely pathogenic variant in an affected patient". | You can add additional unrelated patients (probands) with the same variant: 1. Expand the criterion description by clicking on 2. For Proband 1 (the name can be changed by clicking on the current one and entering a new one), click on the search box and select the variant phase; 3. In the activated search box, select the variant classification; 4. To add another patient, click on .After adding all patients, click on the button that appears to apply the criterion significance level corresponding to the added patients. After that, the criterion state will change to MET (the criterion meets the variant) and the criterion significance level will correspond to the variant classification in added patients. To delete a patient, click on |
| Supporting evidence of benign impact 2 (BP2) | Supporting | Benign | Observed either in trans with a (likely) pathogenic variant for a fully penetrant dominant disorder, or in cis with a (likely) pathogenic variant for a fully penetrant disorder in any inheritance pattern. |
7. In vitro or functional studies#
Functional studies (in vitro or in vivo), considered when applying this section criteria, should be validated, shown to be reproducible and robust in a clinical diagnostic laboratory setting. Do not use in silico studies including protein modelling.
In vitro or functional studies criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Pathogenic Strong 3 (PS3) | Strong | Pathogenic | Functional studies prove a damaging effect of the variant on the gene or gene product. - The criterion significance level depends on strength of evidence (e.g. if only one reference is available, use the criterion with lower significance). Generally, in vivo studies provide stronger evidence thanin vitro studies, and assays that rescue phenotypes provide stronger evidence than assays manipulating them. - If studies provide support at the gene rather than variant level (e.g. biochemical analysis), this should be incorporated within the phenotypic specificity criterion PP4. |
| Strong evidence of benign 3 (BS3) | Strong | Benign | Functional studies show no effect of the variant on the gene or gene product. |
8. Protein-truncating variants#
Protein-truncating variants criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion | Additional options |
| Pathogenic Very Strong 1 (PVS1) | Very strong | Pathogenic | LoF-variants in a gene where loss of function (LoF) is a known mechanism of disease. LoF-variants are genetic variants predicted to severely disrupt protein-coding genes: nonsense, frameshift, canonical ±1 or ±2 splice sites, or initiation codon mutations, or single or multi-exon deletions/duplications. - Do not use in combination with PM4 or PP3. | To use a decision tree to determine the LoF effect of a variant in the context of gene structure and molecular etiology, such as alternative splicing or nonsense-mediated mRNA decay (NMD), click on  . In the window that opens, step by step, select the characteristics that correspond to the variant. You can move between steps . In the window that opens, step by step, select the characteristics that correspond to the variant. You can move between stepsusing  and and  . As a result, you will get a decision: the state and strength (significance level) of the PVS1 criterion, depending on the specific LoF effect of the variant. . As a result, you will get a decision: the state and strength (significance level) of the PVS1 criterion, depending on the specific LoF effect of the variant. |
| Pathogenic Moderate 4 (PM4) | Moderate | Pathogenic | An in-frame deletion/insertion or a stop-loss variant in a non-repeat region of a gene that result in protein length changes. - Do not use in combination with PVS1 or PP3. - Use at supporting level for single amino acid in-frame deletion/insertion. | |
| Supporting evidence of benign impact 3 (BP3) | Supporting | Benign | In-frame deletions/insertions in a repetitive region of a gene. Without a known function. - Do not use in combination with BP4. |
9. Missense variants#
Missense variants criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Pathogenic Strong 1 (PS1) | Strong | Pathogenic | Results in a substitution with the same amino acid change as a previously established variant determined to be pathogenic for this disease. - Generally, used for missense variants, but may also be used at moderate level for initiation codon variants. - Use at supporting level for different intronic change with the same or more severe splicing effect with reference to the predicted impact on the mRNA/protein. - Beware of changes that impact splicing rather than at the amino acid/protein level. |
| Pathogenic Moderate 5 (PM5) | Moderate | Pathogenic | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. - Use at strong level if there are multiple observations of different pathogenic variants at the same residue. - Use at supporting level if the variant is classified as likely pathogenic and there is only one case reported. - Beware of changes that impact splicing rather than at the amino acid/protein level. |
| Pathogenic Moderate 1 (PM1) | Moderate | Pathogenic | Located in a mutational hotspot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation. A mutational hotspot can be considered if pathogenic variants are observed at a high frequency in one or several nearby residues. In silico protein modelling data can be included as supporting evidence. - Use at strong level for very specific residues that are critical for protein structure or function. |
| Supporting evidence of pathogenicity 2 (PP2) | Supporting | Pathogenic | Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease. - Z scores ≥3.09 (marked amber in gnomAD) are significant, but it is important to consider constraint for the region encompassing the variant, not just across the entire gene. Z score is calculated by comparing the expected versus observed number of missense variants. The higher Z score, the higher the constraint or intolerance to variation. |
| Supporting evidence of benign impact 1 (BP1) | Supporting | Benign | Missense variant in a gene for which primarily truncating variants are known to cause disease. - Use the criterion if all or nearly all (e.g., >90%) of the disease-causing variants are truncations. |
10. In silico predictions#
At the top of the section, there are the results of predicting the effect of amino acid substitution on protein function:
- DBNSFP DANN score is an effect prediction score made using DANN;
- DBNSFP MetaLR prediction is an effect prediction value made using Meta LR;
- DBNSFP GERP++ RS is an effect prediction score (rankscore) made using GERP++.
In silico predictions criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Supporting evidence of pathogenicity 3 (PP3) | Supporting | Pathogenic | At least three lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc). Caveat: as many in silico algorithms use the same or very similar input for their predictions, each algorithm should not be counted as an independent criterion. - The criterion can be used only once in any evaluation of a variant. - Do not use in combination with PVS1 or PM4. - Use PP3 if 2/3 tools predict deleterious effect, and use BP4 if 2/3 tools predict benign effect and there is no conservation or 3/3 tools predict benign effect. |
| Supporting evidence of benign impact 4 (BP4) | Supporting | Benign | At least three lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc). Caveat: As many in silico algorithms use the same or very similar input for their predictions, each algorithm should not be counted as an independent criterion. BP4 can be used only once in any evaluation of a variant. - The criterion can be used only once in any evaluation of a variant. - Do not use in combination with BS3. - Use PP3 if 2/3 tools predict deleterious effect, and use BP4 if 2/3 tools predict benign effect and there is no conservation or 3/3 tools predict benign effect. - Use in combination with BP7 for synonymous variants affecting weakly conserved nucleotides which do not impact on splicing. |
| Supporting evidence of benign impact 7 (BP7) | Supporting | Benign | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site, and the nucleotide is not highly conserved. - Use in combination with BP4 for synonymous variants affecting weakly conserved nucleotides which do not impact on splicing. |
11. Literature data#
Literature data criteria#
| Criterion | Criterion significance level | Criterion pathogenicity | Description of the variant that meets the criterion |
| Supporting evidence of benign impact 5 (BP5) | Supporting | Benign | Found in a case with an alternate molecular basis for disease (basis that can explain the patient’s phenotype). - Generally, the criterion is more applicable to dominant disorders with an obvious alternate mechanism. - For recessive disorders, incidental carriers can harbor pathogenic variants unrelated to the disease phenotype. |
| Supporting evidence of pathogenicity 5 (PP5) | Supporting | Pathogenic | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. - The ClinGen Sequence Variant Interpretation (SVI) Working Group (2018) recommends not to use PP5 and BP6 criteria. |
| Supporting evidence of benign impact 6 (BP6) | Supporting | Benign | Reputable source recently reports variant as benign but the evidence is not available to the laboratory to perform an independent evaluation. - The ClinGen Sequence Variant Interpretation (SVI) Working Group (2018) recommends not to use PP5 and BP6 criteria. |
Determine pathogenicity on the "ACMG Classification" tab#
- The first time you open "ACMG Classification" tab, the variant criteria status will be determined automatically.
- You can change the state and significance level of the criteria and add comments to them.
- To erase all results of variant interpretation,
click on
 .
.
note
If pathogenicity was determined as a result of combining the ACMG criteria, then once the results of variant interpretation are erased, the variant is automatically excluded from the sample interpretation results.
- To reset to automatic variant interpretation,
click on
 .
.
As a result of combining the criteria, the variant is classified by pathogenicity (Benign, Likely benign, Uncertain significance, Conflicting interpretations of pathogenicity, Likely pathogenic, Pathogenic) and the interpretation result is set on the pathogenicity scale:
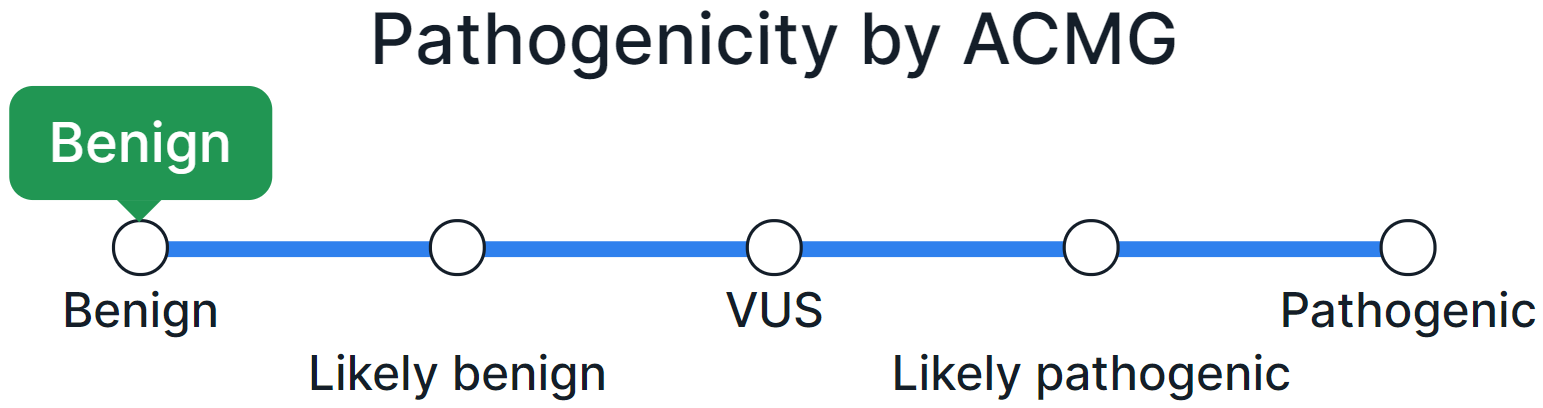
If the pathogenicity class of a variant has been determined by automatic interpretation, a warning to confirm the interpretation will appear.
- To manually classify a variant by pathogenicity, click on the name or point with the value on the pathogenicity scale:

info
The variant pathogenicity class determined manually is stored in the system and will automatically be determined for the same variant in samples annotated later with a warning to confirm the pathogenicity.
info
If a variant has been determined as Pathogenic or Likely pathogenic, it is automatically included to report (if pathogenicity has been confirmed).
Include to interpretation results#
All variants with a certain pathogenicity class are automatically added to the sample interpretation results. If you want to exclude a variant with a determined pathogenicity class from the interpretation results, disable the "Include to interpretation results" option in the upper right corner of the page.
Determine pathogenicity by database#
If you have uploaded a pathogenicity database into the system as described here, the base will annotate sample variants. Thus, if variants are presented in the base, their pathogenicity will correspond to the pathogenicity class in the base (and marked as "manually determined"), but such a pathogenicity needs to be confirmed.
Variant with determined pathogenicity#
After completing the classification of the variant by pathogenicity (according to ACMG or manually) or after annotating the variants by pathogenicity base, the pathogenicity button will change accordingly:
- Pathogenic variant:

- Likely pathogenic variant:

- Variant of uncertain significance:

- Variant with conflicting interpretations of
pathogenicity:

- Likely benign variant:

- Benign variant:

info
All variants with a certain pathogenicity class are automatically added to the sample interpretation results. If you want to exclude a variant with a determined pathogenicity class from the interpretation results, disable the "Include to interpretation results" option in the pathogenicity dialog or on the variant details page.
info
If a variant has been determined as Pathogenic or Likely pathogenic, it is automatically included to report (if pathogenicity has been confirmed).
The way of determining the variant pathogenicity ("by ACMG" or "manually determined") can be found in the tooltip by hovering over the pathogenicity button.
Variants can be sorted from pathogenic to benign by clicking on "Pathogenicity" column name once, and from benign to pathogenic by clicking on the column name twice.
Variants can be filtered by pathogenicity:
- "Pathogenicity" filter in advanced filtering mode;
- an additional filter "Pathogenicity" in basic filtering mode (can be added on "Profile" page).
Clear pathogenicity#
You can remove the result of the classification of a variant by pathogenicity by clicking on the pathogenicity button in the variant row and selecting "Clear" option in the dialog:
![]()
info
Once pathogenicity has been cleared, the variant is automatically excluded from the sample interpretation results.
Confirm pathogenicity#
Confirmation of the pathogenicity class of a variant may be required in the following cases:
- The automatic variant pathogenicity classification by ACMG was restored after any manipulations with the criteria on "ACMG Classification" tab;
- Variant annotation or earlier sample analysis stages have been reprocessed;
- The pathogenicity class of the variant was obtained from the pathogenicity base.
- The pathogenicity class of the variant was stored in the system after manual determination of pathogenicity.
On SNV Viewer page, variants that require pathogenicity confirmation are
indicated by ![]() next to the pathogenicity button.
If you agree with the variant pathogenicity, click on the bell or the pathogenicity button and
select "Confirm Pathogenicity" option in the dialog:
next to the pathogenicity button.
If you agree with the variant pathogenicity, click on the bell or the pathogenicity button and
select "Confirm Pathogenicity" option in the dialog:

On "ACMG Classification" tab, the confirmation warning looks like this:

If you agree with the variant pathogenicity,
click on  .
.
info
Once pathogenicity is confirmed, the variant is automatically added to the sample interpretation results. If you want to exclude a variant from interpretation results, disable the "Include to interpretation results option in the pathogenicity dialog or on the variant details page.
info
A variant can be added to the sample interpretation results without confirming the variant pathogenicity. To do this, enable the “*Include to interpretation results” option on the variant details page.