Advanced Filtering of Variants
To enter the Advanced Variant Filtering mode, click
on the  button at the top of the table.
You will see the Query Builder:
button at the top of the table.
You will see the Query Builder:

note
If you are on the page of SNV Viewer with somatic mutations, then the somatic mutation filtering condition ("Origin in Somatic") will be added by default:

info
On "Profile Settings" page, you can adjust which filtering mode you will see when you first open sample SNV Viewer page: basic or advanced.
note
Table filtering is saved when you exit the SNV Viewer page.
Filtering Columns#
The columns are divided into groups:
- Common
- Per-sample data (if there is data on sequencing samples)
- Gene
- Affected transcripts
- gnomAD 3
- gnomAD 4
- ClinVar
- Conservation
- ExAC
- Protein function effect
- Protein function effect (additional)
- Other frequencies
- Other
- Custom annotation sources (if at least one custom annotation was added to the system before the variants in the sample were annotated)
You can interact with column groups by
using ![]() and
and ![]() to
expand and collapse the column list of the group, respectively.
to
expand and collapse the column list of the group, respectively.
Use the search box to find the filtering column. The search is case-insensitive. Some columns can be found by search synonyms. For example, the following columns can be found by the word "change":

Common#
- Sample Status (basic filter "Sample Status") - filter variants by run sample interpretation status (completed or uncompleted). Single value filter. Only available in run SNV Viewer.
- Interpretation - filter variants by interpretation (comment) added by the user on the variant details page or panel. Any value filter.
- HPO Phenotype - filter variants by terms from Human Phenotype Ontology (HPO).
To open the phenotype search window, click on the filter value
field
 . How to find and select the
phenotypes in this form is described here.
. How to find and select the
phenotypes in this form is described here. - Gene panel (basic filter "Gene panel") - filter variants by localization in certain genes from the panel. Single value filter. You can add a gene panel on on "Gene Panels" page or import a panel from the Library of the most common clinical panels (such option is available for basic filter "Gene panel" in basic filtering mode. If you don't have any gene panels added, the list of filter values will be empty.
- Region (basic filter "Region") - filter variants
by position on the genome (chromosome, start and end positions). To filter by region, click on the input
window
 and enter the position of the
needed genomic interval, i.e. chromosome, start and end positions. Possible options of writing the interval
position:
and enter the position of the
needed genomic interval, i.e. chromosome, start and end positions. Possible options of writing the interval
position:
chr1:161,190,824-161,193,921chr1:161190824-161193921chr1:161190824_1611939211:161190824-161193921
You can also filter by more than one interval at once. For example,
by chr1:161190824-161193921, chrX:151110769-151113866.
To complete adding the condition, press Enter or click outside edit value container.
The filtering result will be variants whose positions intersect with the entered interval.
- Gene (column "Locus", basic filter "Gene") - filter variants by localization in gene transcript. The filter values depend on the selected filter operator:
- any value if the operators "starts with", "contains", "not contains" are selected. For example, this is useful if you want to filter variants by genes, the name of which contains a certain letter sequence (e.g., "HLA-").
- multiselect if the operators “in” and “not in” are selected. Used to filter variants by localization in certain genes. In the value options, the gene transcript is given in brackets next to the official gene name.
- no value if the operators "not empty", "empty" are selected.
- Impact - filter by the predicted effect of the variant on the protein: High, Moderate, Low, Modifier. Column values are described here. Multiselect filter.
- Genome position - filter by variant position in the genome: in exon (Exonic), in intron (Intronic), in the intergenic region (Intergenic). Multiselect filter.
- Consequence (basic filter "Consequence") - filter by the effect of the variant on genes. Column values are described here. Multiselect filter.
- Pathogenicity (column "Pathogenicity", basic filter "Pathogenicity") - multiselect filter by variant pathogenicity, which have been determined either manually, by ACMG criteria, or according to the pathogenicity base.
- Label (column "Label", basic filter "Label") - multiselect filter by variant significance defined by the user: High significance, Moderate significance, Low significance, Sequencing error, Undefined.
- Phasing group (column "PG") - multiselect filter by phasing group status:
- Phasing group - merging of homozygous variants located within a specific genomic interval;
- Part of group consequence equal - part of phasing group with concordant amino acid substitutions;
- Part of group consequence unequal - part of phasing group with discordant amino acid substitutions.
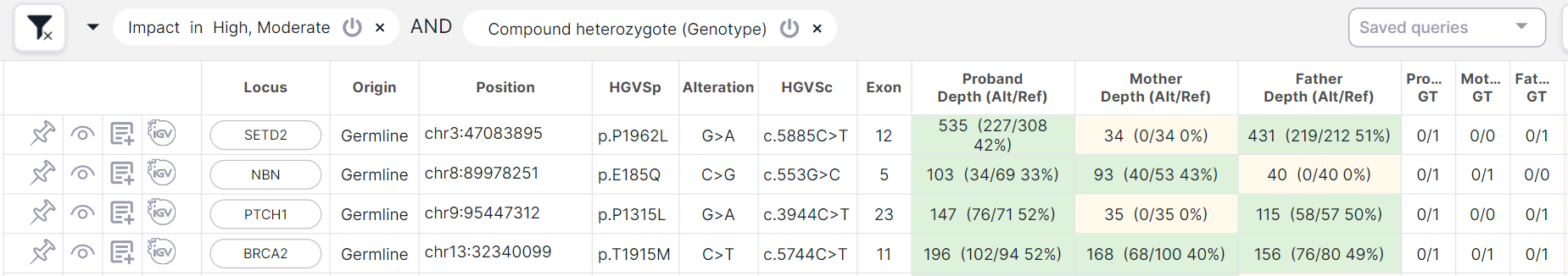
- Compound heterozygote (Genotype) - filter variants by whether they are compound heterozygotes (i.e. heterozygosity of two or more variants of the same gene; all cases are described in detail here). Unlike the compound heterozygote filtering mode, this filter is applied simultaneously with other filtering conditions. As a result, the variants included in the compound heterozygote will be found, but the remaining variants from this compound heterozygote may be filtered out by other conditions.
In family analysis SNV Viewer and in SNV Viewer with one sequencing sample, to filter variants by compound heterozygote, select the filter in the list of filtering columns:

In germline cohort analysis SNV Viewer, in SNV Viewer with several sequencing samples and in run SNV Viewer (if there is data on sequencing samples), to filter variants by compound heterozygote, select a sample in which all cases will be found where at least two heterozygotes (0/1, 1/0, 0|1, etc.) are located in the same gene:

- Trio GT (Genotype) - multiselect filter by variant inheritance: De novo variants, Newly formed homozygotes (homozygotes in proband that are heterozygous in parents) or Single parent-inherited. Only available in family group analysis for Trio case (Proband, Father and Mother).
- Chromosome (column "Position")- filter variants by localization in chromosomes: chr1-chr22, chrX, chrY, chrM, chromosomes like chrN_random (contigs located in the N chromosome but could not be reliably ordered within the sequence, or sequences for alternative haplotypes), chromosomes like chrUn_ (contigs that cannot be confidently placed on a specific chromosome), chrEBV. Multiselect filter.
- rsId (column "External links") - filter variants by ID from dbSNP database. Filter with ID input. You can use as a value either the full ID (e.g. rs1126497) or the ID digits only (e.g. 1126497).
- ClinVar ID (column "External links") - filter variants by Variation ID from ClinVar database (e.g. 183700). Filter with ID input.
- Origin (basic filter "Origin") - filter variants by mutation type: Somatic, Germline. Multiselect filter.
- Start (column "Position") - filter by the start position of the variant on the chromosome. Numeric filter (natural number).
- End - filter by the end position of the variant on the chromosome. Numeric filter (natural number).
- Reference allele (column "Alteration") - filter by reference allele of the variant. Any value filter.
- Alternative allele (column "Alteration") - filter by alternative allele of the variant. Any value filter.
- HGVSc - filter variants by nucleotide substitution using the HGVS notation: “c.” (coding; for a substitution in the coding sequence) or “n.” (non-coding; for a substitution in the non-coding sequence) prefix + genomic position of the substituted nucleotide + reference allele > alternative allele. Any value filter.
- HGVSp (short) (column "HGVSp") - filter variants by amino acid substitution using the HGVS notation in the single-letter amino acid code (“p.” prefix (protein) + reference amino acid + amino acid position in protein + new amino acid resulting from the substitution). Any value filter. e.g.: p.P72R.
- HGVSp (long) (column "HGVSp") - filter variants by amino acid substitution using the HGVS notation in the three-letter amino acid code. Written with transcript: Ensembl transcript ID + “p.” prefix (protein) + reference amino acid + amino acid position in protein + new amino acid resulting from the substitution. Any value filter. e.g.: ENSP00000269305.4:p.Pro72Arg.
- Exon - filter by exon number in the transcript that the variant affected. Numeric filter (natural number).
- Onco Relevance - filter by the variant significance for any oncological disease: Relevant, Irrelevant. Single value filter.
- Caller Filters - multiselect filter by variant quality (GATK filters).
- Occurrence in Run - filter variants by occurrence in run samples. Numeric filter (integer number). Only available in run SNV Viewer and run sample SNV Viewer.
- Cosmic ID - filter by variant ID in COSMIC database (e.g. COSV52665487). Filter with ID input. Only available if COSMIC database has been uploaded as a custom annotation before the sample variant annotation is complete.
- RUSeq AF - filter variants by alternative allele frequency from RUSeq database. Numeric filter (any number). Only available if RUSeq database has been uploaded as a custom annotation before the sample variant annotation is complete.
Per-sample data#
- The filtering column group is only present if there is sequencing sample data.
- Genotype (column "GT") - filter variants
by genotype, i.e. reference and alternative allele values for the sample. Genotypes are filtered ignoring phasing
information. To add a condition for filtering variants by genotype, do the following:
1. Select a sample to filter:
- "Some": any sample has the required genotype;
- "Every": each sample has the required genotype;
- Sample name.
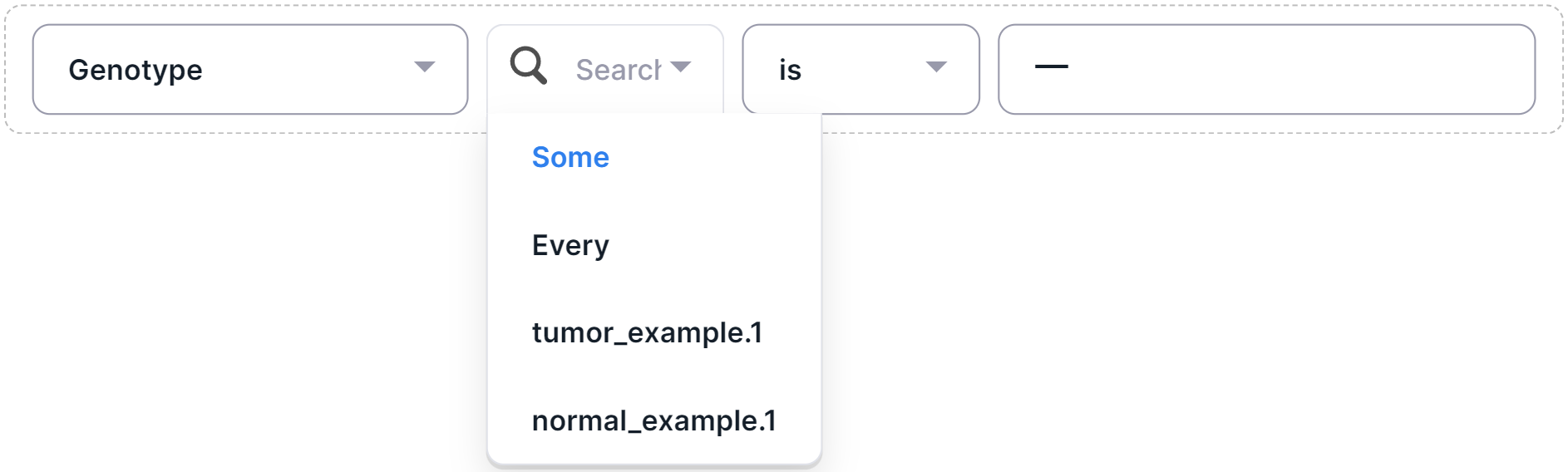
- Select a filter operator:
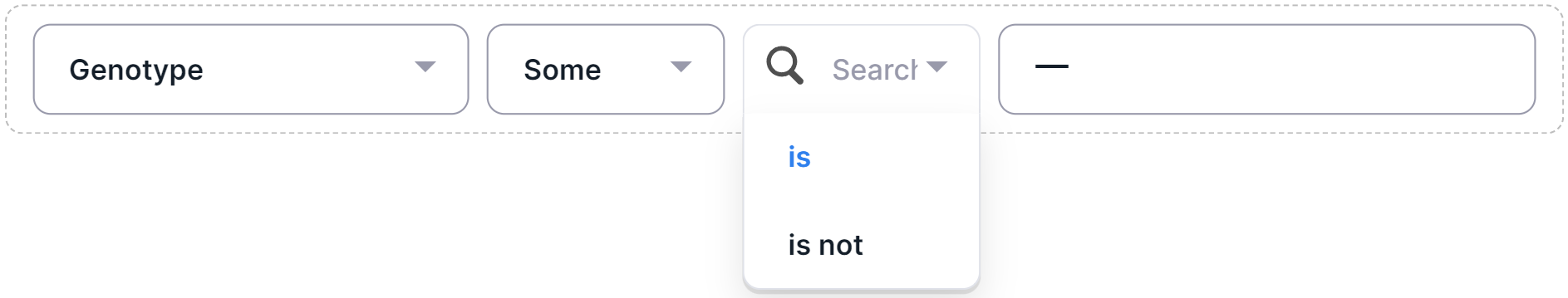
- Select the required filter value by clicking on the value row:
- "REF": only reference allele at the site, (e.g.,
0/0,0|0,0/0/0); - "ALT": there is at least one alternative allele at the site (e.g.,
1/1,1|1,0/1,1/0,0|1,1|0,1/.,1|.,./1,.|1,1,0/0/1,0/2,1/2); - "HOM": homozygous site, can be as in reference or alternative (e.g.,
1|1,1/1,1/1/1,0/0,0|0); - "HET": heterozygous site, containing at least two different alleles (e.g.,
0/1,1/0,0|1,1|0,2/0,0/1/2); - "HOM_ALT": homozygous site in alternative (e.g.,
1/1,1|1|1); - "MIS": information is missing for the site (e.g.,
./.).
- Read depth (column "Depth (Alt/Ref)") - filter variants by sequencing depth, i.e. the total number of reads of the sequence overlapping the variant position for this sample.
- Ref count - filter variants by the number of times the reference nucleotide was read in the sequence for this sample.
- Alt count - filter variants by the number of times the alternative nucleotide was read in the sequence for this sample.
- AF - filter variants by alternative allele frequency for this sample.
To add a condition for filtering variants by depth, ref, alt or AF, do the following:
- Select a filter function:
- "Max": maximum value;
- "Min": minimum value;
- "Avg": mean;
- "Sum": total value.

- Select a filter operator:
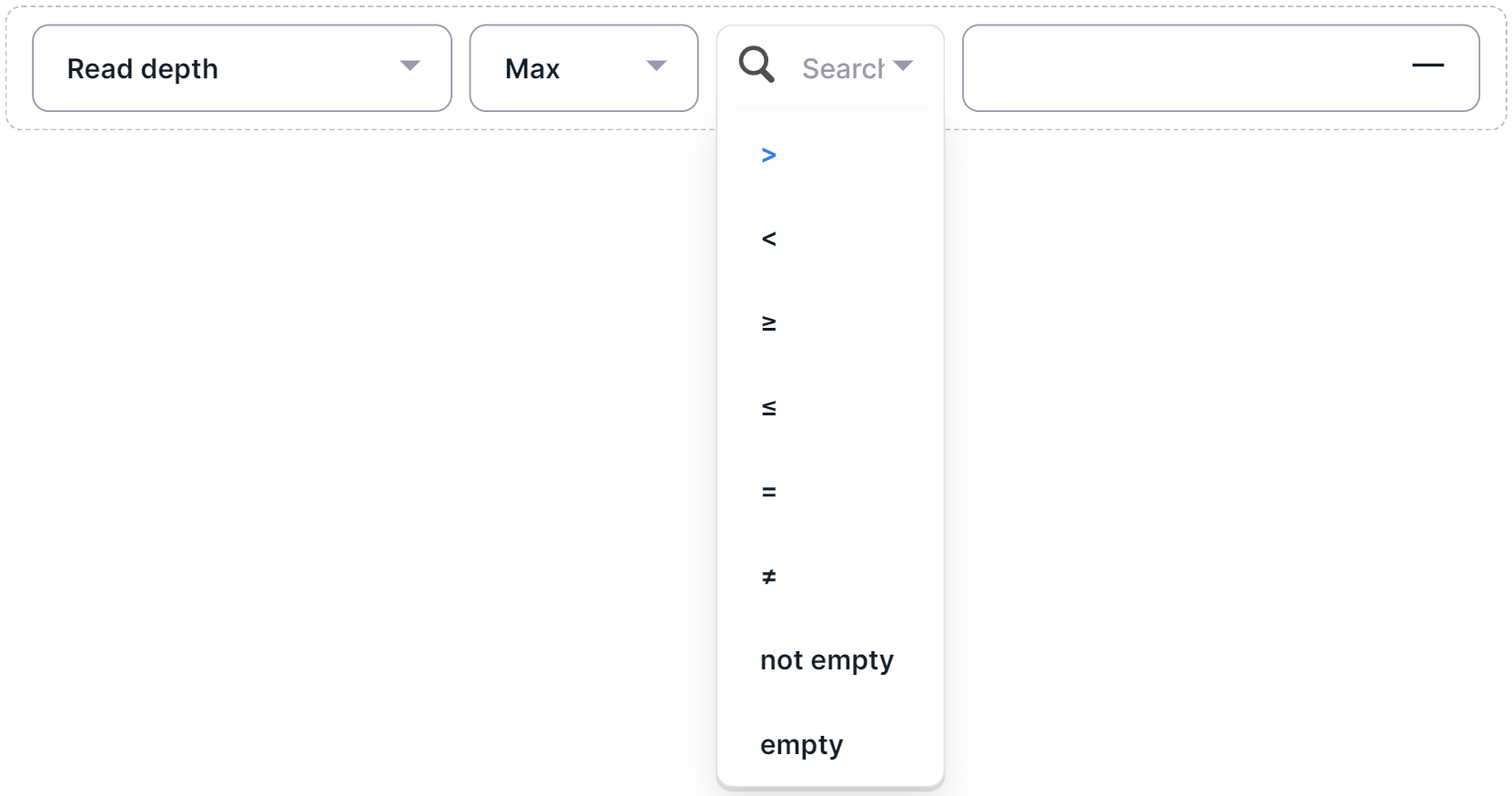
- If a mathematical operator was selected as the filter operator, click on input
box
 and enter the filter threshold
(natural number for filters by depth, ref and alt count, and positive rational number for AF).
To complete adding the condition, press Enter or click outside edit value container.
and enter the filter threshold
(natural number for filters by depth, ref and alt count, and positive rational number for AF).
To complete adding the condition, press Enter or click outside edit value container.
Gene#
- RefSeq Transcript - filter variants by transcript ID in RefSeq database (e.g. NM_002354.3). Any value filter.
- OMIM ID - filter variants by gene ID in OMIM database (e.g. 185535). Filter with ID input.
- NCBI ID - filter variants by gene ID in NCBI Gene database (e.g. 4072). Filter with ID input.
- Swiss-Prot ID - filter variants by gene ID in UniProtKB Swiss-Prot database (e.g. P16422). Filter with ID input.
- TrEMBL ID - filter variants by gene ID in UniProtKB TrEMBL database (e.g. B5MCA4). Filter with ID input.
- Ensembl ID - filter variants by gene ID in Ensembl database (e.g. ENSG00000119888). Filter with ID input.
- pLI - filter variants by pLI (Probability of Loss-of-Function Intolerance) - the probability that a transcript is intolerant to loss-of-function (LoF) variants. Range: 0 to 1. Genes with pLI ≥ 0.9 are considered LoF-intolerant. Numeric filter (positive rational number).
- LOEUF - filter variants by LOEUF (Loss-of-function Observed/Expected Upper bound Fraction) - the upper bound of the 90% confidence interval for the observed/expected ratio of high-confidence pLoF (predicted loss-of-function) variants. Lower values (LOEUF < 0.35) indicate a gene is more constrained against LoF variants. Numeric filter (positive rational number).
- Missense O/E ratio - filter variants by the upper bound of the 90% confidence interval for the observed/expected ratio of missense variants. Lower values suggest a gene is more constrained against missense variation. Numeric filter (positive rational number).
- Missense Z-score - filter variants by a Z-score representing the deviation of the observed number of missense variants in a transcript from the expected number under a model of neutral mutation distribution. Greater positive values (Z-score ≥ 3.09) indicate that the transcript is more intolerant of missense variation. Lower or negative Z-scores indicate that the gene tolerates more missense mutations than expected. Numeric filter (rational number).
Affected transcripts#
The group includes the Is MANE transcript filter, which allows filtering variants based on whether they affect the gene's main transcript according to MANE Select, MANE plus clinical, or Ensembl canonical.
The filter is implemented as a toggle switch:
- Turn the switch on to include only variants that affect the gene’s main transcript:
![]()
- Turn the switch off to filter for variants that do not affect the gene’s main transcript:
![]()
Phenotypes#
- OMIM Phenotype - filter by the names of OMIM phenotypes associated with the gene in which the variant is located. Any value filter. Only available if you have access to OMIM database data inside Genomenal, which is provided if the user has a license to access the database.
- OMIM Inheritance - filter by the mode of inheritance of OMIM phenotypes. Multiselect filter. Only available if you have access to OMIM database data inside Genomenal, which is provided if the user has a license to access the database.
Possible modes of inheritance:
- Autosomal dominant (AD);
- Autosomal recessive (AR);
- Digenic dominant (DD);
- Digenic recessive (DR);
- Inherited chromosomal imbalance (ICB);
- Isolated cases (IC);
- Mitochondrial (Mi);
- Multifactorial (Mu);
- Somatic mosaicism (SMo);
- Somatic mutation (SMu);
- X-linked (XL);
- X-linked dominant (XLD);
- X-linked recessive (XLR);
- Y-linked (YL);
- Pseudoautosomal dominant (PD);
- Pseudoautosomal recessive (PR);
- Not specified - the phenotype has an empty value in the "Inheritance" column in the OMIM database.
- GenCC Phenotypes - filter by the names of diseases in GenCC database associated with the gene in which the variant is located. Any value filter.
gnomAD 3#
Filter columns with the following data from gnomAD v3 database:
- AF (column "gnomAD 3 AF") - filter variants by the total allele frequency (AF). Numeric filter (positive rational number).
- AC - filter variants by the total alternative allele count in high quality genotypes. Numeric filter (natural number).
- AN - filter variants by the total number of called high quality genotypes. Numeric filter (natural number).
- XY AF - filter variants by allele frequency (AF) in the XY subpopulation. Numeric filter (positive rational number).
- XX AF - filter variants by allele frequency (AF) in the XX subpopulation. Numeric filter (positive rational number).
- Number of Homozygotes - filter variants by the number of homozygotes. Numeric filter (natural number).
- [Population] AF - filter variants by total allele frequency (AF) in different populations, where [Population] is Ashkenazi Jewish, Amish, African and Afr. Am., Finnish, South Asian, Latino, Non-finnish european, East Asian, Middle Eastern, Other. Numeric filter (positive rational number).
- [Population] XY/XX samples AF - filter variants by allele frequencies (AF) in XY/XX subpopulations in different populations, where [Population] is Ashkenazi Jewish, Amish, African and African American, Finnish, South Asian, Latino, Non-finnish european, East Asian, Middle Eastern, Other. Numeric filter (positive rational number).
- [Population] Number of Homozygotes - filter variants by the number of homozygotes in different populations, where [Population] is Ashkenazi Jewish, Amish, African and African American, Finnish, South Asian, Latino, Non-finnish european, East Asian, Middle Eastern, Other. Numeric filter (positive rational number).
- Coverage - filter variants by average coverage depth by bases: <10, 10-100, ⩾100. Any value filter.
gnomAD 4#
Filter columns with the following data from gnomAD v4 database:
- AF - filter variants by the total alternative allele frequency (AF) in high quality genotypes in exomes and genomes. Numeric filter (positive rational number).
- AC - filter variants by the total alternative allele count in exomes and genomes. Numeric filter (natural number).
- AN - filter variants by the total number of called high quality genotypes in exomes and genomes. Numeric filter (natural number).
- Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in exomes and genomes. Numeric filter (natural number).
- XY Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XY subpopulation in exomes and genomes. Numeric filter (natural number).
- XX Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XX subpopulation in exomes and genomes. Numeric filter (natural number).
- XY AF - filter variants by the total alternative allele frequency in the XY subpopulation in exomes and genomes. Numeric filter (positive rational number).
- XY AC - filter variants by the total alternative allele count in high quality genotypes in the XY subpopulation in exomes and genomes. Numeric filter (natural number).
- XY AN - filter variants by the total number of called high quality genotypes in the XY subpopulation in exomes and genomes. Numeric filter (natural number).
- XX AF - filter variants by the total alternative allele frequency in the XX subpopulation in exomes and genomes. Numeric filter (positive rational number).
- XX AC - filter variants by the total alternative allele count in high quality genotypes in the XX subpopulation in exomes and genomes. Numeric filter (natural number).
- XX AN - filter variants by the total number of called high quality genotypes in the XX subpopulation in exomes and genomes. Numeric filter (natural number).
- Exomes AF - filter variants by the total alternative allele frequency (AF) in high quality genotypes in exomes. Numeric filter (positive rational number).
- Exomes AC - filter variants by the total alternative allele count in exomes. Numeric filter (natural number).
- Exomes AN - filter variants by the total number of called high quality genotypes in exomes. Numeric filter (natural number).
- Exomes Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in exomes. Numeric filter (natural number).
- Exomes XY Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XY subpopulation in exomes. Numeric filter (natural number).
- Exomes XX Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XX subpopulation in exomes. Numeric filter (natural number).
- Exomes XY AF - filter variants by the total alternative allele frequency in the XY subpopulation in exomes. Numeric filter (positive rational number).
- Exomes XY AC - filter variants by the total alternative allele count in high quality genotypes in the XY subpopulation in exomes. Numeric filter (natural number).
- Exomes XY AN - filter variants by the total number of called high quality genotypes in the XY subpopulation in exomes. Numeric filter (natural number).
- Exomes XX AF - filter variants by the total alternative allele frequency in the XX subpopulation in exomes. Numeric filter (positive rational number).
- Exomes XX AC - filter variants by the total alternative allele count in high quality genotypes in the XX subpopulation in exomes. Numeric filter (natural number).
- Exomes XX AN - filter variants by the total number of called high quality genotypes in the XX subpopulation in exomes. Numeric filter (natural number).
- Genomes AF - filter variants by the total alternative allele frequency (AF) in high quality genotypes in genomes. Numeric filter (positive rational number).
- Genomes AC - filter variants by the total alternative allele count in genomes. Numeric filter (natural number).
- Genomes AN - filter variants by the total number of called high quality genotypes in genomes. Numeric filter (natural number).
- Genomes Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in genomes. Numeric filter (natural number).
- Genomes XY Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XY subpopulation in genomes. Numeric filter (natural number).
- Genomes XX Number of Homozygotes - filter variants by the total number of individuals homozygous for alternative allele in the XX subpopulation in genomes. Numeric filter (natural number).
- Genomes XY AF - filter variants by the total alternative allele count in high quality genotypes in the XY subpopulation in genomes. Numeric filter (natural number).
- Genomes XY AC - filter variants by the total alternative allele count in high quality genotypes in the XY subpopulation in genomes. Numeric filter (natural number).
- Genomes XY AN - filter variants by the total number of called high quality genotypes in the XY subpopulation in genomes. Numeric filter (natural number).
- Genomes XX AF - filter variants by the total alternative allele count in high quality genotypes in the XX subpopulation in genomes. Numeric filter (natural number).
- Genomes XX AC - filter variants by the total alternative allele count in high quality genotypes in the XX subpopulation in genomes. Numeric filter (natural number).
- Genomes XX AN - filter variants by the total number of called high quality genotypes in the XX subpopulation in genomes. Numeric filter (natural number).
ClinVar#
Filter columns with the following data from ClinVar database:
- Clinical significance (column "ClinVar", basic filter "ClinVar") - filter variants by the clinical significance (you can find the description of values here). Multiselect filter.
- Allele ID - filter variants by allelic substitution ID (e.g. 405185). Filter with ID input.
- Phenotypes - filter variants by the phenotypic significance. Any value filter.
- Phenotypes cross references - filter variants by phenotype references in various databases (Orphanet, MedGen, MONDO, OMIM, etc.) Any value filter.
- Haplotype phenotypes - filter by the phenotypic significance of the variant that is part of the haplotype. Any value filter.
- Haplotype phenotypes cross references - filter by references to the phenotype of a variant that is part of the haplotype in various databases (Orphanet, MedGen, MONDO, OMIM, etc.) Any value filter.
- Review status - filter variants by the summary score of the data source, in which the clinical significance of the phenotype is declared. Any value filter. Possible value options and their descriptions can be found here.
- Cross references - filter variants by references in various databases (ClinGen, UniProtKB, OMIM, GTR, etc.) Any value filter.
- Allele origin - filter variants by allele origin. Multiselect filter.
- Variant Haplotype ID - filter by ID of the variant (Variation ID) that is part of the haplotype. Filter with ID input.
Conservation#
- Ancestral allele - filter variants by ancestral allele predicted by EPO. Any value filter.
- Altai Neanderthal - filter variants by Altai Neanderthal genotype. Any value filter.
- Denisova - filter variants by Denisova genotype. Any value filter.
- Vindija Neanderthal - filter variants by Vindija Neanderthal genotype. Any value filter.
- [Program] [Score] - filter variants by conservation scores obtained using prediction by various programs, where [Program] is a prediction program: FitCons, GERP++, PhyloP, PhastCons, SiPhy, BStatistic, and [Score] is a conservation score: rankscore, score, neutral rate. Numeric filter (positive rational number).
- [Score] confidence value - filter variants by confidence value of various conservation scores: Highly significant; Significant; Informative; Other, where [Score] is a conservation score: Integrated, GM12878, H1-hESC, HUVEC. Multiselect filter.
- [Distribution] - filter variants by stationary allele distribution in the site, estimated using SiPhy algorithm, where [Distribution] is a distribution of alleles A, G, C or T: pA, pG, pC, pT. Numeric filter (positive rational number).
ExAC#
Filter columns with the following data from ExAC database:
- AF - filter variants by the total allele frequency (AF). Numeric filter (positive rational number).
- Allele count - filter variants by alternative allele count. Numeric filter (natural number).
- Adjusted AF - filter variants by allele frequency (AF) in a population with genotype quality ≥20 and depth ≥10. Numeric filter (positive rational number).
- Adjusted allele count - filter variants by alternative allele count in a population with genotype quality ≥20 and depth ≥10. Numeric filter (natural number).
- [Population] AF - filter variants by total allele frequency (AF) in different populations, where [Population] is African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian. Numeric filter (positive rational number).
- [Population] allele count - filter variants by alternative allele count in different populations, where [Population] is African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian. Numeric filter (positive rational number).
- [Cohort] AF - filter variants by total allele frequency (AF) in different cohorts, where [Cohort] is Non-TCGA, Non-Psych. Numeric filter (positive rational number).
- [Cohort] Allele count - filter variants by alternative allele count in different cohorts, where [Cohort] is Non-TCGA, Non-Psych. Numeric filter (natural number).
- [Cohort] adjusted AF - filter variants by allele frequency (AF) in a population with genotype quality ≥20 and depth ≥10 in different cohorts in ExAC database, where [Cohort] is Non-TCGA, Non-Psych. Numeric filter (positive rational number).
- [Cohort] adjusted allele count - filter variants by alternative allele count in a population with genotype quality ≥20 and depth ≥10 in different cohorts, where [Cohort] is Non-TCGA, Non-Psych. Numeric filter (natural number).
- [Cohort] [Population] AF - filter variants by total allele frequency (AF) in different populations in different cohorts, where [Cohort] is Non-TCGA, Non-Psych, and [Population] is African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian. Numeric filter (positive rational number).
- [Cohort] [Population] allele count - filter variants by alternative allele count in different populations in different cohorts,where [Cohort] is Non-TCGA, Non-Psych, and [Population] is African & African American, American, East Asian, Finnish, Non-Finnish European, South Asian. Numeric filter (natural number).
Protein function effect#
- [Algorithm] [Score] - filter variants by prediction scores of the effect of amino acid substitution on protein function, obtained using various algorithms and programs, where [Algorithm] is a prediction algorithm: SIFT, Polyphen, SIFT4G, LRT, FAtHMM, Mutation Accessor, and [Score] is a prediction score: rankscore, score, omega, p-value. Numeric filter (positive rational number).
- [Algorithm] prediction - filter variants by the results of predicting the effect of amino acid substitution on protein function, obtained using various algorithms and programs, where [Algorithm] is a prediction algorithm: Polyphen, SIFT4G, LRT, FAtHMM, Mutation Accessor. Multiselect filter.
- SpliceAI Delta score - filter by the probability of the variant being splice-altering at any position within a given interval (+/-50 bp between the variant and gained/lost splice site by default), predicted SpliceAI. Numeric filter (any number).
- SpliceAI Delta score (acceptor gain) - filter by the probability of the variant being splice acceptor at any position within a given interval (+/-50 bp between the variant and gained/lost splice site by default), predicted SpliceAI. Numeric filter (any number).
- SpliceAI Delta score (acceptor loss) - filter by the probability of the variant not to be splice acceptor at any position within a given interval (+/-50 bp between the variant and gained/lost splice site by default), predicted SpliceAI. Numeric filter (any number).
- SpliceAI Delta score (donor gain) - filter by the probability of the variant being splice donor at any position within a given interval (+/-50 bp between the variant and gained/lost splice site by default), predicted SpliceAI. Numeric filter (any number).
- SpliceAI Delta score (donor loss) - filter by the probability of the variant not to be splice donor at any position within a given interval (+/-50 bp between the variant and gained/lost splice site by default), predicted SpliceAI. Numeric filter (any number).
- SpliceAI Delta position - filter by the location (in bp) where splicing changes relative to the variant position (positive values are downstream of the variant, negative values are upstream). Numeric filter (integer number).
- SpliceAI Delta position (acceptor gain) - filter by the location (in bp) of splice acceptor relative to the variant position (positive values are downstream of the variant, negative values are upstream). Numeric filter (integer number).
- SpliceAI Delta position (acceptor loss) - filter by the location (in bp) which is not a splice acceptor relative to the variant position (positive values are downstream of the variant, negative values are upstream). Numeric filter (integer number).
- SpliceAI Delta position (donor gain) - filter by the location (in bp) of splice donor relative to the variant position (positive values are downstream of the variant, negative values are upstream). Numeric filter (integer number).
- SpliceAI Delta position (donor loss) - filter by the location (in bp) which is not a splice donor relative to the variant position (positive values are downstream of the variant, negative values are upstream). Numeric filter (integer number).
- CADD raw - filter by the extent to which the annotation profile for a variant suggests that the variant is likely to be "observed" (negative values) vs "simulated" (positive values), calculated by CADD. Numeric filter (any number).
- CADD phred - filter by Phred-scaled score of the deleteriousness of a variant, calculated by CADD. Numeric filter (any number).
- MaxEntScan Alt score - filter by score for alternative allele predicting the loss of a native splice site for a variant, calculated by MaxEntScan. Numeric filter (any number).
- MaxEntScan Ref score - filter by score for reference allele predicting the loss of a native splice site for a variant, calculated by MaxEntScan. Numeric filter (any number).
- MaxEntScan diff - filter by the difference between the scores for alternative and reference alleles predicting the loss of a native splice site for a variant, calculated by MaxEntScan. Numeric filter (any number).
Protein function effect (additional)#
- [Algorithm] [Score] - filter variants by prediction scores of the effect of amino acid substitution on protein function, obtained using various algorithms and programs, where [Algorithm] is a prediction algorithm: BayesDel addAF, BayesDel noAF, Provean, Meta SVM, Meta LR, M-CAP, MutPred, MVP, MPC, Primate AI, DEOGEN2, DANN, FAtHMM-MKL, FAtHMM-XF, Eigen, and [Score] is a prediction score: rankscore, score, p-value. Numeric filter (positive rational number).
- [Algorithm] prediction - filter variants by the results of predicting the effect of amino acid substitution on protein function, obtained using various algorithms and programs, where [Algorithm] is a prediction algorithm: BayesDel addAF, BayesDel noAF, Provean, Meta SVM, Meta LR, M-CAP, Primate AI, FAtHMM-MK, FAtHMM-XF. Multiselect filter.
- Transcript ID: filter variants by transcript ID in UniProtKB database (e.g. P16422) for which the effect of amino acid substitution on protein function has been predicted by MutPred. Any value filter.
- Amino acid change - filter variants by amino acid substitution whose effect on protein function was predicted by MutPred. Any value filter. e.g.: G35r.
- Top 5 features - filter variants by the top five substitution effects on protein function predicted by MutPred. Any value filter.
Other frequencies#
- 1000G Allele frequency - filter variants by total allele frequency in the 1000 Genomes Project. Numeric filter (positive rational number).
- 1000G [Population] AF - filter variants by total allele frequency (AF) in different populations in the 1000 Genomes Project, where [Population] is East Asian, European, African, American, South Asian, African American, European American. Numeric filter (positive rational number).
- UK10K allele frequency - filter variants by total allele frequency in combined genotypes in the UK10K cohort from the UK10K project. Numeric filter (positive rational number).
- UK10K allele count - filter variants by alternative allele count in combined genotypes in the UK10K cohort from the UK10K project. Numeric filter (natural number).
- UK10K [Cohort] AF - filter variants by total allele frequency (AF) in combined genotypes in different cohorts from the UK10K project, where [Cohort] is TwinsUK, ALSPAC. Numeric filter (positive rational number).
- UK10K [Cohort] allele count - filter variants by alternative allele count in combined genotypes in different cohorts from the UK10K project, where [Cohort] is TwinsUK, ALSPAC. Numeric filter (natural number).
- ESP [Population] AF - filter variants by total allele frequency (AF) in different populations in NHLBI GO Exome Sequencing Project (ESP), where [Population] is African American, European American. Numeric filter (positive rational number).
- [Population] allele count - filter variants by alternative allele count in different populations from NHLBI GO Exome Sequencing Project (ESP), where [Population] is African American, European American. Numeric filter (natural number).
Other#
- Coding sequence position - filter variants by coding sequence position. Numeric filter (natural number).
- Protein position - filter variants by protein position. Numeric filter (natural number).
- Amino acids - filter variants by reference amino acid. Any value filter.
- Codons - filter variants by nucleotide substitution written as codons. Any value filter. e.g.: GAA/GAG.
- Amino acid position - filter variants by amino acid position. Numeric filter (natural number).
- APPRIS annotation - filter variants by human splicing isoforms (APPRIS annotation): Principal 1-5; Alternative 1-2. Multiselect filter.
- Geuvadis EQTL target gene - filter variants by gene from eQTL data analysis in GEUVADIS. Any value filter.
- ENIGMA clinical significance - filter by clinical significance of the variant located in the known or suspected breast and/or ovarian predisposition genes (such as BRCA1, BRCA2) determined by ENIGMA consortium. Multiselect filter: Pathogenic, Likely pathogenic, Benign, Likely benign, Not provided.
- ENIGMA clinical significance comment - filter by comment on clinical significance of the variant located in the known or suspected breast and/or ovarian predisposition genes (such as BRCA1, BRCA2) determined by ENIGMA consortium. Any value filter.
Custom annotation sources#
A filtering column group is present if at least one custom annotation was added to the system before the sample variants were annotated. Filtering column names look like this: [custom annotation name]: [annotation column name]. To add a condition for filtering variants by column from a custom annotation, do the following:
- Select a filter operator. If the annotation column type is text, then the operators will be the following:

If the annotation column type is numeric, then the operators will be the following:

- If operators other than "not empty" and "empty" have been selected, click on the input
box and enter the filter column value.
To complete adding the condition, press Enter or click outside edit value container.
Compound Heterozygote Filtering Mode#
A compound heterozygote is a genetic condition in which two or more alleles of the same gene carry different mutations in a patient. This condition is observed if the patient inherits a mutant allele from each parent or inherits one mutation from one of the parents, and the second occurs de novo. Compound heterozygotes are common in autosomal recessive diseases.
If there is data about sequencing samples, a button for filtering by compound heterozygote appears in the query builder:
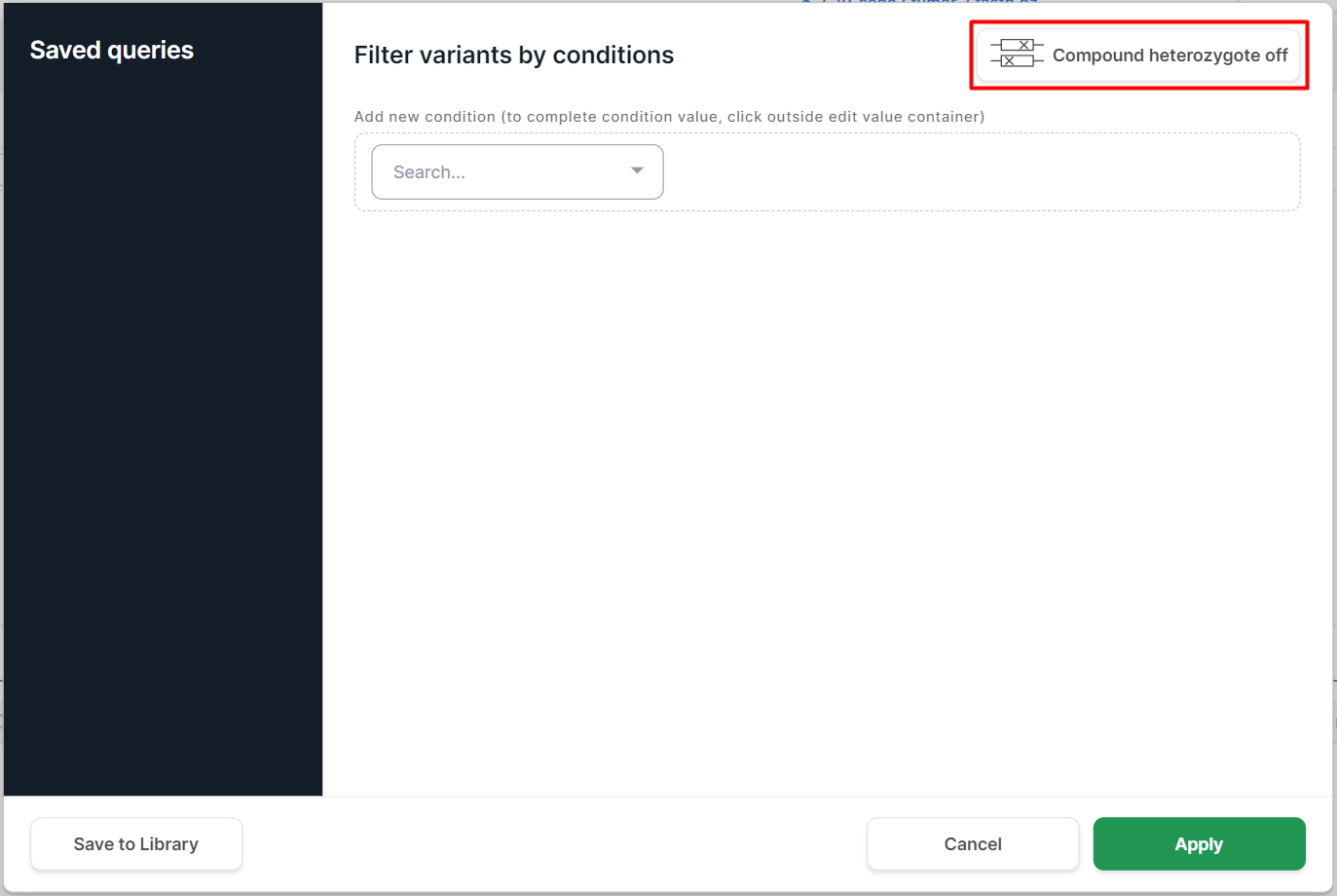
Filtering by compound heterozygote depends on SNV Viewer type:
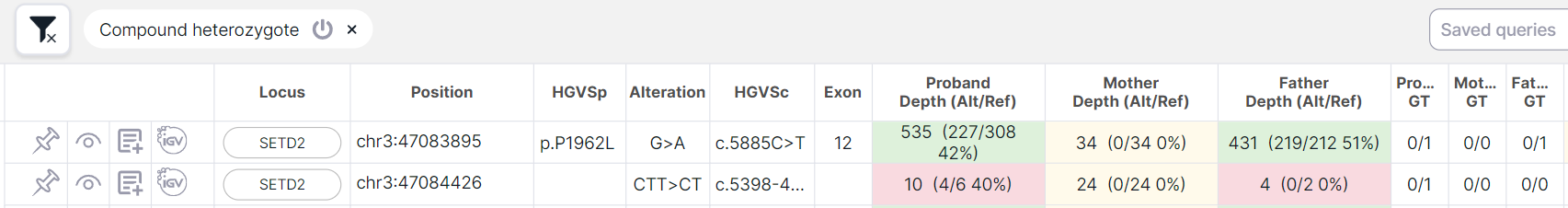
SNV Viewer of family trio analysis (Proband, Father, Mother): the proband has at least two heterozygotes in one gene (0/1, 1/0, 0|1, etc.) with the following inheritance options:
- at least one of the heterozygotes is inherited from the father, and at least one is inherited from the mother:
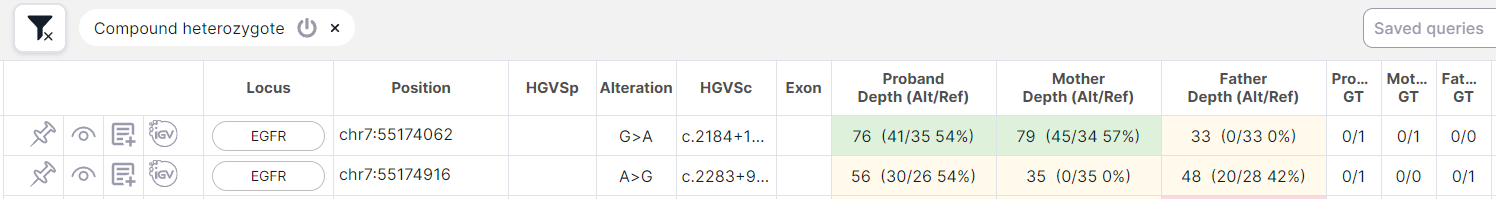
- at least one of the heterozygotes occurred de novo (i.e the father and mother have homozygous reference 0/0), and the second is inherited from the father or from the mother:
To apply a filter by compound heterozygote, click the
button  . As a result, there will be found all cases where at least two heterozygotes are located in the same gene in the proband, and in the parents they are either heterozygous, or homozygous reference, or there is no information about the site (./.). To turn off the filter in the query builder, click the button
. As a result, there will be found all cases where at least two heterozygotes are located in the same gene in the proband, and in the parents they are either heterozygous, or homozygous reference, or there is no information about the site (./.). To turn off the filter in the query builder, click the button  .
.
SNV Viewer of family duo analysis (Father and Mother): cases when in one gene at least one heterozygote (0/1, 1/0, 0|1, etc.) is in the mother and at least one is in the father:

To apply a filter by compound heterozygote, click the
button , and to turn off the filter in the query builder, click the button .
SNV Viewer of family duo analysis (Proband, Parent (Father or Mother)): the proband has at least two heterozygotes in one gene (0/1, 1/0, 0|1, etc.) with the following inheritance options:
- at least one heterozygote is inherited, and the second is not inherited from the parent (i.e. the parent has homozygous reference 0/0):
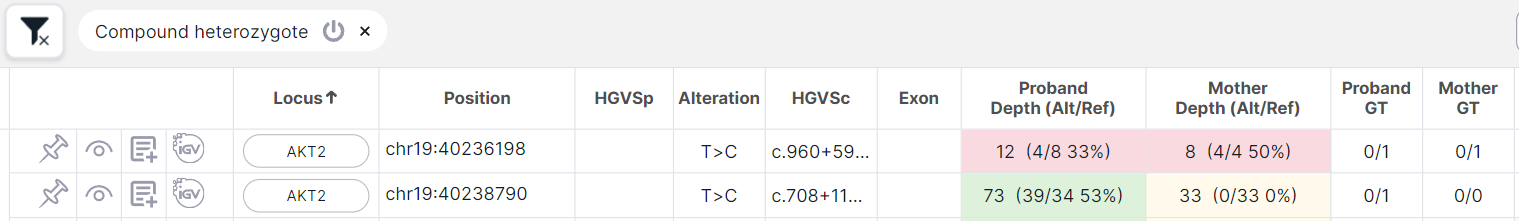
- both are not inherited from the parent (i.e. the parent has homozygous reference 0/0):
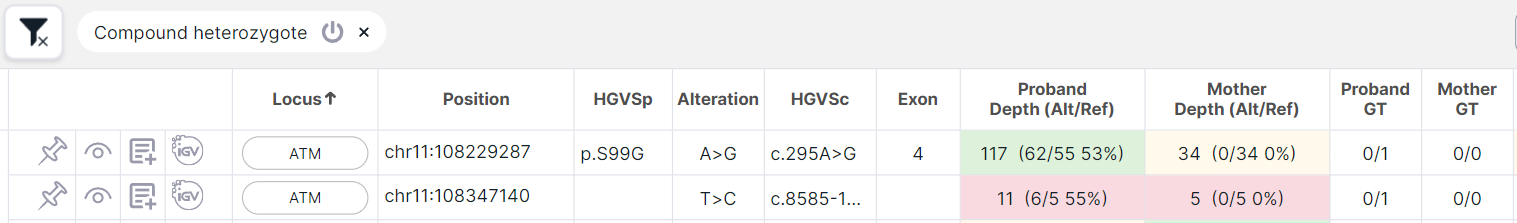
To apply a filter by compound heterozygote, click the
button , and to turn off the filter in the query builder, click the button .
SNV Viewer with one sequencing sample (e.g. for a single sample of tumor or healthy tissue): there will
be found all cases where at least two heterozygotes (0/1, 1/0, 0|1, etc.) are located in the same gene.
To apply a filter by compound heterozygote, click the
button , and to turn off the filter in the query builder, click the button .
SNV Viewer of germinal cohort analysis or SNV Viewer with several sequencing samples (e.g. for a tumor/control sample pair), or run SNV Viewer (if there is data on sequencing samples): by clicking on the compound heterozygote filter button, you will see a drop-down sample list:

When one of the samples is selected, the variants will be filtered in such a way that in this sample there will be at least two heterozygotes in one gene (0/1, 1/0, 0|1, etc.) To turn off such a filter in the query builder, you need to click on the compound heterozygote filter button and select "Turn off" option.
Applying of the compound heterozygote filtering mode with other filtering conditions:
When applying the compound heterozygote filtering mode together with other filtering conditions, the condition for compound heterozygote is already applied to those variants that fit the other conditions:
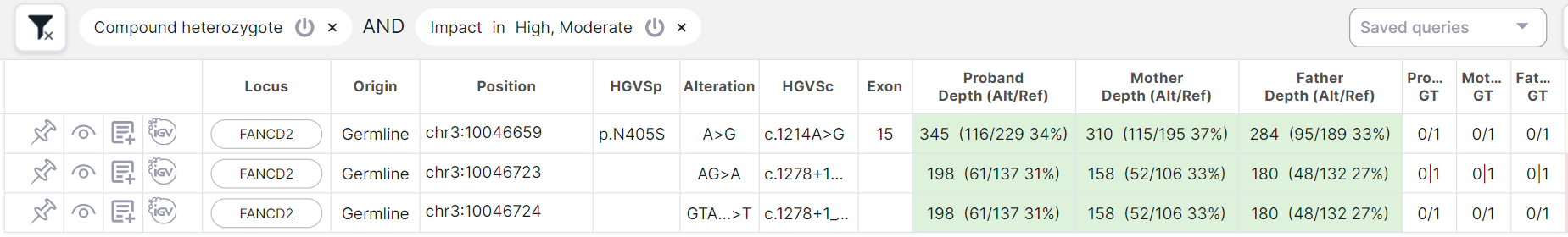
This fundamentally differs the filtering mode from "Compound heterozygote (Genotype)" filter, which is applied simultaneously with other filtering conditions. As a result of applying the filter, there will be found variants that are included in the compound heterozygote, but the remaining variants from this compound heterozygote can be filtered out by other conditions:
Add the First Filter Condition#
To add a new condition, click on the highlighted window:

You will see a list of annotation columns available for filtering variants:

The way of adding a filter condition depends on the filter type:
1. Multiselect Filters#
- Ability to select multiple column values at once.
- The way of adding a filter condition:
- Select a filter column from the list of all columns;
- Select a filter operator:
- "in";
- "not in";
- "not empty": the selected column has a non-empty value in the annotation for the searched variants (there is no such option for the "Chromosome" and "Origin" columns);
- "empty": the selected column has no value in the annotation for the searched variants (there is no such option for the "Chromosome" and "Origin" columns).
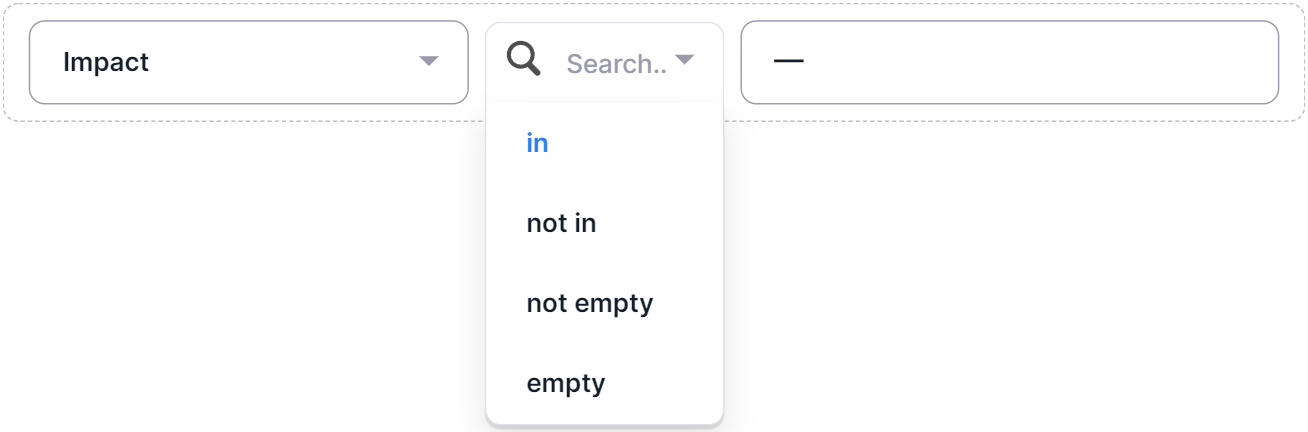
- If the "in" or "not in" operators have been selected, select the required filter column values by clicking on the value rows:
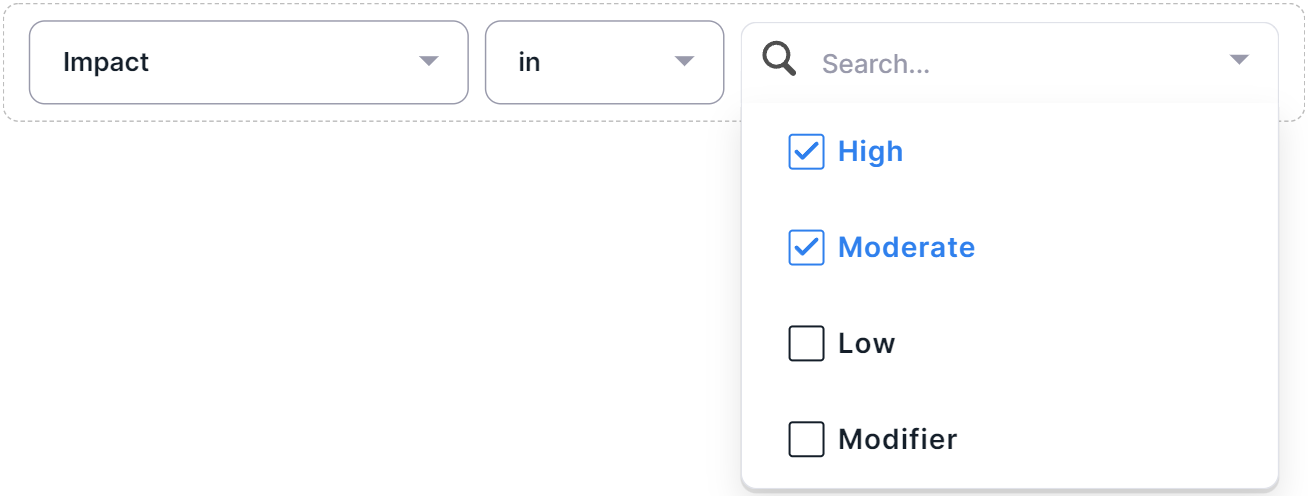
- To complete adding the condition, click outside edit value container.
2. Single Value Filters#
- Ability to select only one filter value.
- The way of adding a filter condition:
- Select filter from the list of all filters. The filter operator "is" is automatically selected for filters of this type;
- Select the required filter value by clicking on the value row:

3. Filters with ID input#
- Ability to enter the required column value.
- The way of adding a filter condition:
- Select a filter column from the list of all columns;
- Select a filter operator:

- If the operators "is" or "is not" were selected, click on the input
box
 and enter filter column value:
and enter filter column value:

You can enter several IDs at once in a list separated by a comma (e.g. rs1234, rs2799066, rs4648562), a semicolon
or a space (rs1234 rs2799066 rs4648562).
- To complete adding the condition, press Enter or click outside edit value container.
4. Numeric Filters#
- Ability to enter the required column value.
- The way of adding a filter condition:
- Select a filter column from the list of all columns;
- Select a filter operator:
- ">";
- "<";
- "≥";
- "≤";
- "=";
- "≠";
- "not empty": the selected column has a non-empty value in the annotation for the searched variants (there is no such option for the "Start" and "End" columns);
- "empty": the selected column has no value in the annotation for the searched variants (there is no such option for the "Start" and "End" columns).
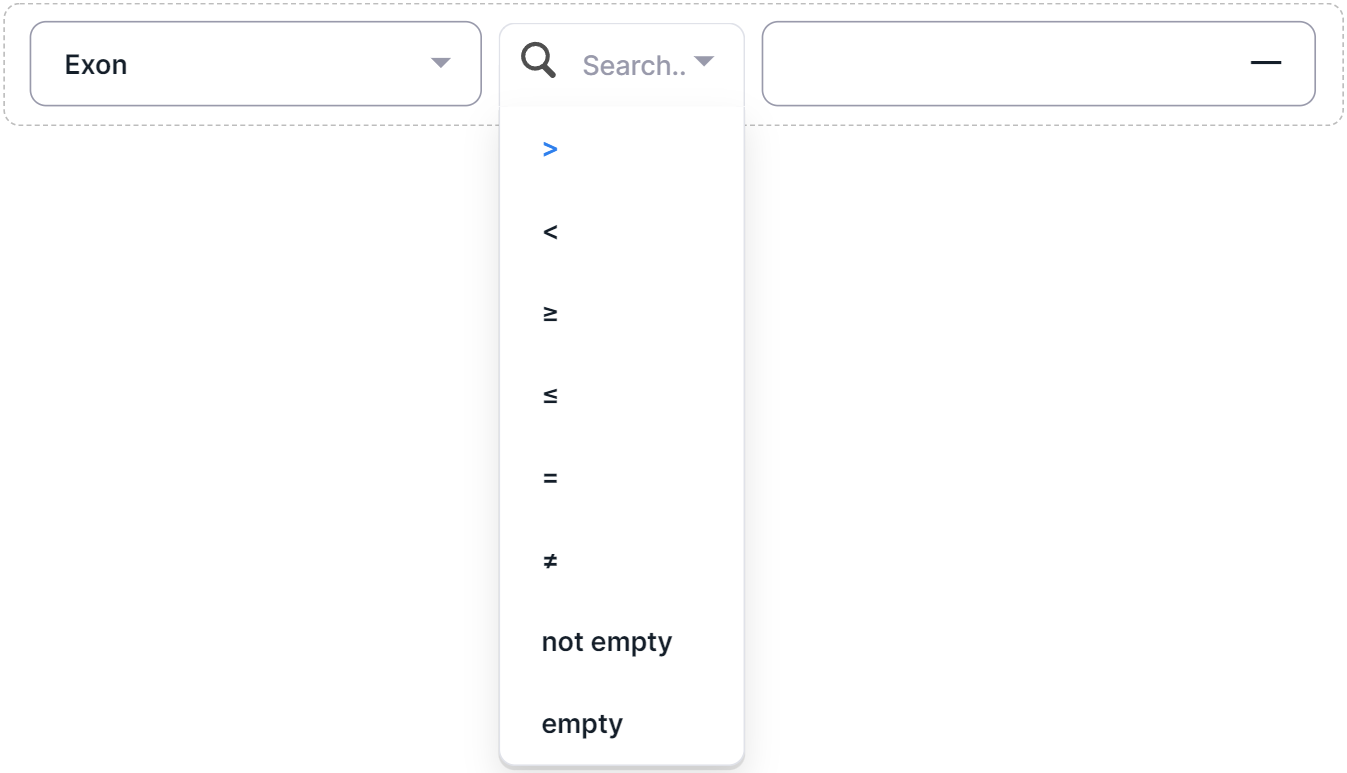
- If a mathematical operator was selected as the filter operator, click on the input
box and enter filter column value:

- To complete adding the condition, press Enter or click outside edit value container.
5. Any Value Filters#
- Ability to enter the required column value.
- The way of adding a filter condition:
- Select a filter column from the list of all columns;
- Select a filter operator:
- "starts with" (there is no such option for the "HGVSc" column);
- "contains";
- "not contains" (there is no such option for the "HGVSc" column);
- "not empty": the selected column has a non-empty value in the annotation for the searched variants (there is no such option for the "Reference allele" column);
- "empty": the selected column has no value in the annotation for the searched variants (there is no such option for the "Reference allele" column).

- If the operators "starts with", "contains" or "not contains" were selected, click on the
input
box and
enter filter column value:

- To complete adding the condition, press Enter or click outside edit value container.
Actions with a Condition in the Query Builder#
In the query builder, you can edit, turn off, or remove an already created condition.
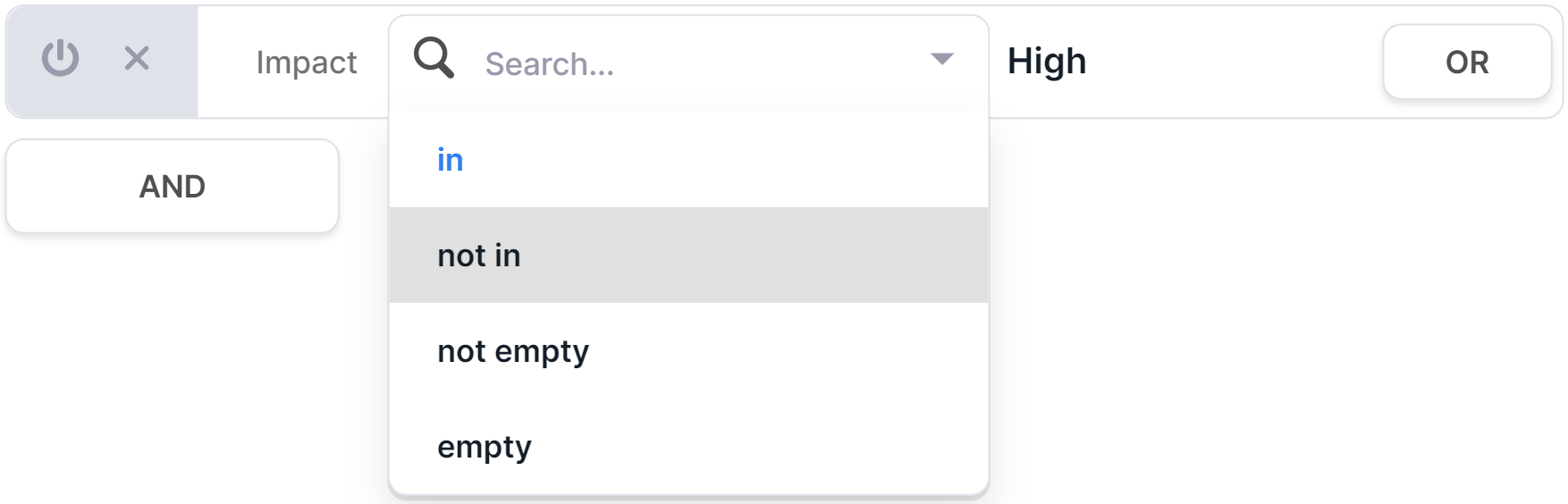
Edit a Condition#
You can change the operator and/or value in the added condition. To do this, you need to click on the corresponding field:

and choose another option:
Turn off a Condition#
Turning off a condition is useful if you don’t want to lose the added condition, but at the moment you
don’t need to filter by it. To turn off a condition, hover over it and
click on ![]() :
:

Remove a Condition#
To remove a condition, hover over it and
click on ![]() :
:
tip
A condition that has not yet been filled in can also be removed (if it's not the very first condition in the Query Builder):

Add the Following Condition#
If you want to add another condition to the query, first decide whether you want the variant you are looking for to strictly match both conditions (the one already added and the new one), or whether the variant can match one of the conditions.
The searched variant matches both conditions#
- After adding the first condition, click on
 .
. - Complete the condition as described in the corresponding section. To get started, click on the highlighted window:

The searched variant matches one of the conditions#
- After adding the first condition, hover over it and
click on
 .
. - Complete the condition as described in the corresponding section. To get started, click on the highlighted window:
Apply a Query#
After filling in the conditions for the searched variants,
click on  .
The query builder window will then close, and the SNV Viewer table will display only those variants
that satisfy the conditions of the applied query.
.
The query builder window will then close, and the SNV Viewer table will display only those variants
that satisfy the conditions of the applied query.
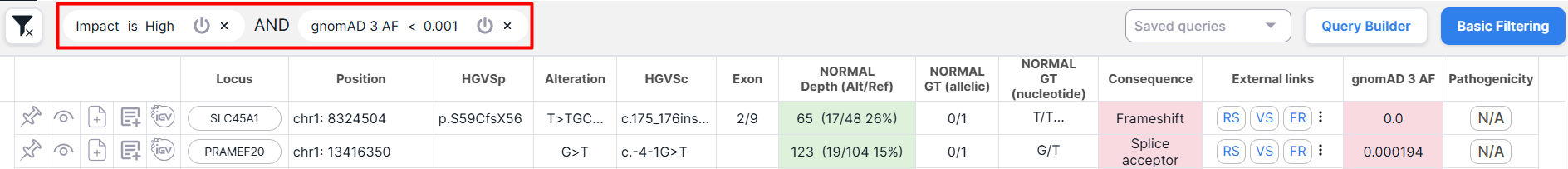
The conditions of the applied query are presented above the table with variants:
To edit, save or create a new query,
open the Query Builder by clicking on  .
.
To return to the SNV Viewer main page,
click on  .
.
Actions with the Conditions of the Applied Query#
The conditions of the applied query are presented above the table with variants:
You can perform the following actions with query conditions:
- Edit the condition value inplace - see below for details.
- Turn the condition off - deactivate without removing it from the query. To do this, click on the button
 . A turned off condition is indicated by a strikethrough:
. A turned off condition is indicated by a strikethrough:

To turn the condition back on, click the same button.
- Remove the condition by clicking on the cross
 .
. - Clear all the applied conditions by clicking on
 .
. - Collapse the conditions. This is useful if the query includes several conditions, resulting in it taking up a lot of space on the page:

To collapse the query conditions, click on
the button ![]() to the right of
the Clear all filters button. Then only a few conditions of the query will be shown:
to the right of
the Clear all filters button. Then only a few conditions of the query will be shown:

To expand the query conditions back, click on ![]() .
.
info
On Profile Settings page, you can adjust whether the query conditions are expanded or collapsed immediately after applying a filter query.
Editing the Condition Value Inplace#
Conditions with the values (that is, those whose operators are not "empty" or "not empty") can be edited inplace above the table of variants. To do this, click on the current value:

and make the changes:
- For multiselect filters: a searchable list of values will open. To select an additional value, check its box. To exclude a value, uncheck its box.
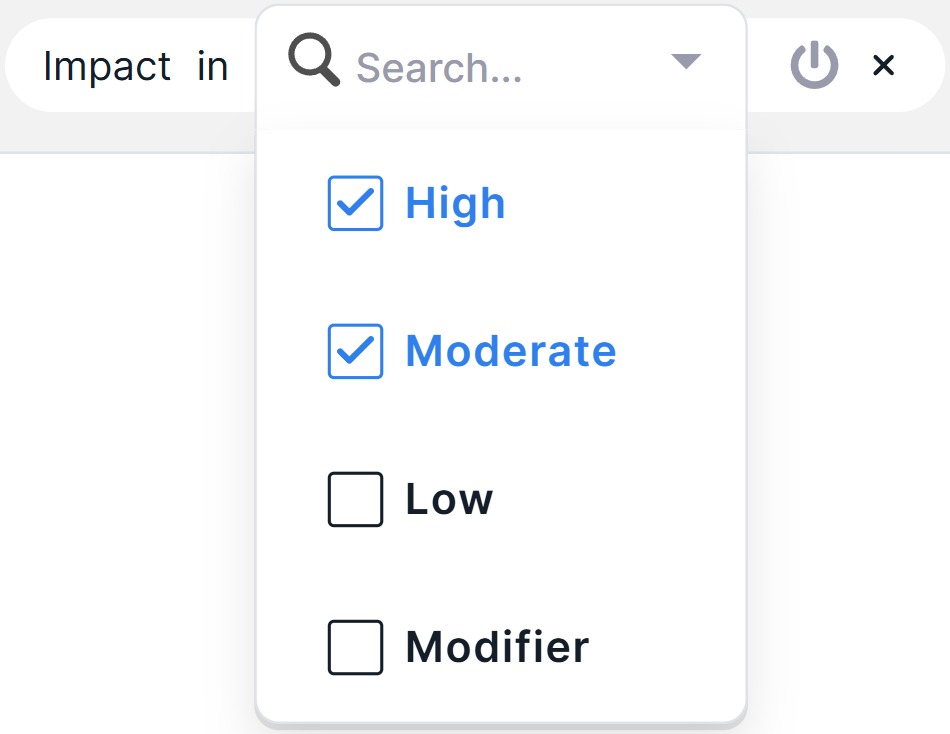
To finish selecting condition values, click in the empty area on the page.
- For single value filters: select a value from the list that opens by clicking on the row:
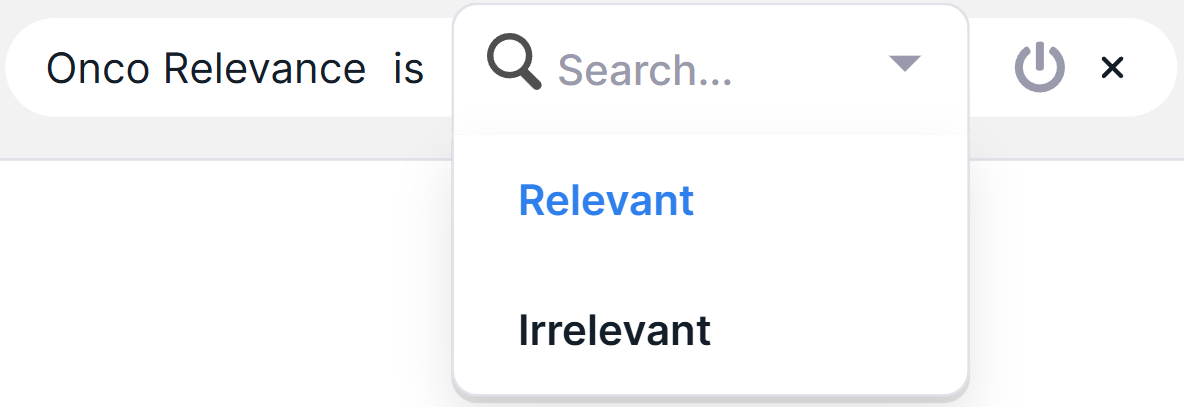
- For filters with value input: edit the value and press Enter or click in the empty area on the page.

All other actions to edit the condition, such as selecting a different condition operator, are performed in the query builder.
Save a Query#
If you want to reuse a set of filtering conditions, save it to the library.
To do this, open the query builder and click
the  button.
In the dialog that opens, enter a query name:
button.
In the dialog that opens, enter a query name:

and click  .
.
Saved queries are displayed in the query builder on the left:

They are also available in the corresponding basic filter options.
To delete a saved query, select it and click the
trash icon ![]() .
.
Filtering by saved queries#
Filtering is available in two modes:
- Single query. To apply a single query, select it in the Saved queries list
and click :
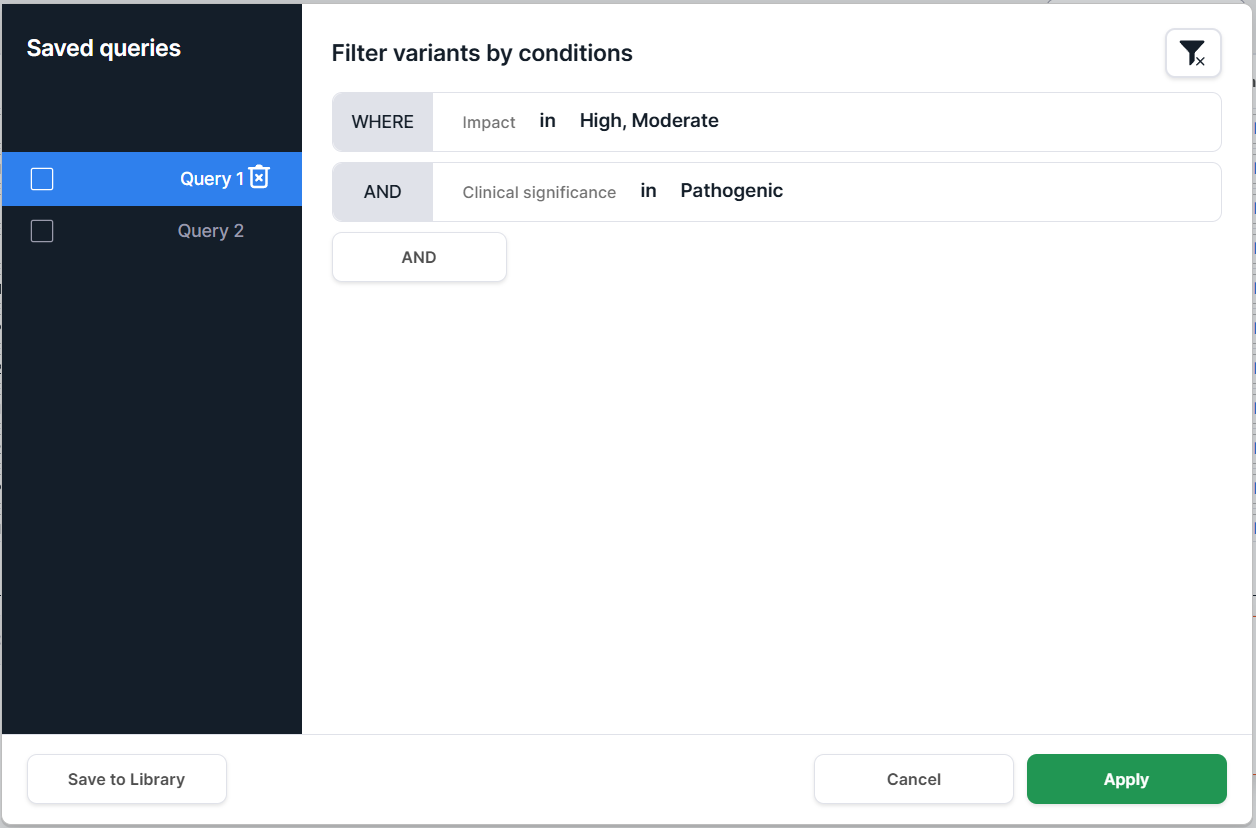
After applying the filter, all conditions included in the selected query are displayed above the table:

When query conditions are modified, an asterisk appears next to the query name:

To save changes:
Click
in the query builder;Then:
- Click to update the existing query;
- Or enter a new name and
click
 to save it as a new query.
to save it as a new query.
- Click
Multiple queries. To apply multiple queries at once, select them using checkboxes and click
 :
:
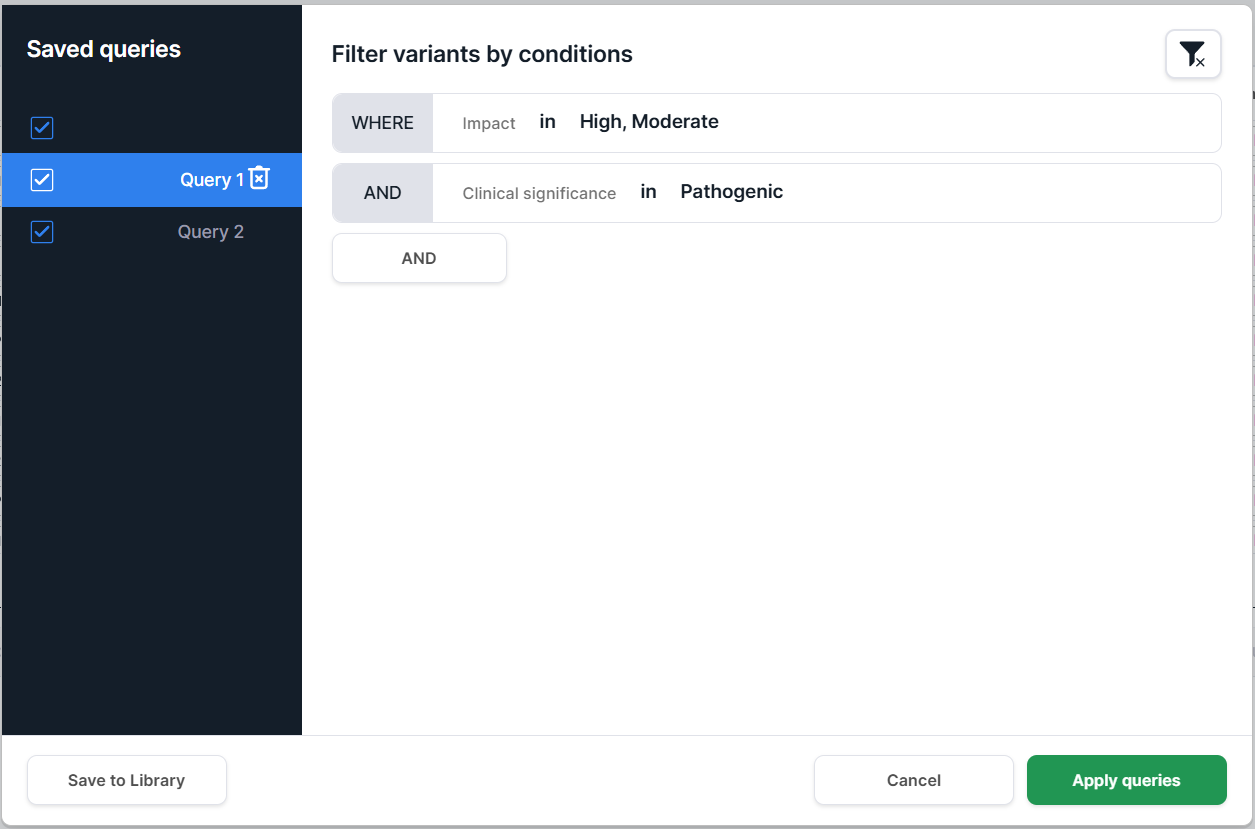
In this mode, filtering is performed as a logical OR combination of the selected queries. Only the query names are displayed above the table. To view the conditions included in a query, open it in the query builder.

Limitations of the multiple queries mode:
- Additional conditions that are not part of saved queries cannot be added to the current filter. To use extra conditions, first save them as a separate query, then select and apply it together with the others.
- When editing a query, the Apply queries button becomes unavailable. If you do not want to save the changes, click Cancel (the changes will not be applied). To apply modified conditions, first save them as an existing or new query, then select it and click Apply queries.
- In this mode, clicking a query does not add it to the filter. It is only used to view the query conditions. To apply a query, you must select its checkbox.